
Abstract
Three recent anti-amyloid-β antibody trials for Alzheimer’s disease reported similar effect sizes, used non-reactive saline as placebo, and showed large numbers of adverse events including imaging anomalies (ARIA) that correlate with cognitive changes. Conversely, all previous antibody trials were less reactive and pronounced ineffective. We argue that these observations point to unblinding bias, inflating apparent efficacy and thus altering the risk-benefit balance. Further, we highlight data demonstrating that beyond reducing amyloid, monoclonal antibodies increase monomeric amyloid-β42 in cerebrospinal fluid, which may explain potential benefits. We should recalibrate the efficacy of these antibodies and devote more resources into strategies beyond removing amyloid.
Of more than 40 Phase 2 and Phase 3 trials targeting amyloid-β (Aβ) in early Alzheimer’s disease (AD), only the latest two met statistical significance for cognitive benefits: lecanemab [1] and donanemab [2]. Both of these agents are potent anti-Aβ monoclonal antibodies (mAbs). Multiple publications have scrutinized potential methodological biases and concerns over risk-benefit ratios [3–5], in particular the significant correlation between the occurrence of amyloid related imaging abnormalities (ARIA) and positive changes on the Clinical Dementia Rating Scale–Sum of Boxes (CDR-SB) [6]. Digma, Winer, and Greicius [6] also point out that the variability of cognitive and clinical effect sizes among mAbs depending on the type of scale used in the trials, and the fact that virtually all mAbs increase the monomeric 42 amino-acid isoform of Aβ (Aβ42) in the cerebrospinal fluid (CSF). We submit that these previously undiscussed issues on efficacy should shape the conversation regarding the risk-benefit tradeoffs of administering mAbs.
THE SCALE MATTERS
The mean treatment effect of donanemab versus placebo on Mini-Mental State Examination (MMSE; range, 0–30), a rater-independent objective scale of cognitive performance, was 0.48 points (95% confidence limits, 0.08 to 0.87 points), representing a mean slowing of progression of 14.8% versus placebo. We calculated the Cohen’s d value for donanemab (Table 2 of Sims et al. [2]), finding d = 0.30 and 0.29 for the CDR-SB scale models NCS2 and MMRM, 0.19 and 0.18 correspondingly for ADAS-Cog13, which contains more objective tests, and 0.14 and 0.12 for MMSE. The Cohen’s d results are comparable to those estimated by Goldberg et al. [7] for the CDR-SB scale (0.23) in suggesting a small effect size. We additionally show that the more objective scales give smaller apparent effect size, supporting the concern that placebo unblinding inflates the reported efficacy.
Unlike the MMSE, the CDR-SB is a semi-structured interview between the rater, the patient, and caregiver. The discrepancy in donanemab effect sizes between these two scales suggests that the true effect of donanemab is very small. An interview-based endpoint can be influenced by the occurrence of intervention-related adverse events. A mean slowing of progression by donanemab of 14.8% on MMSE is at odds with a mean reduction in brain Aβ burden of 85.3% from baseline (–88.0% versus placebo), raising serious questions about the causal relationship between amyloid removal and cognitive improvement. In agreement with its minimal effect on MMSE, donanemab did not decrease plasma levels of neurofilament light chain, a validated biomarker of neuroaxonal damage.
DOES ARIA INFLATE EFFICACY?
ARIA are abnormalities seen on magnetic resonance imaging (MRI) in about 40% of patients treated with humanized mAbs presenting either as edema (ARIA-E) and/or hemorrhage (ARIA-H) [8]. While most meta-analyses of anti-amyloid trials showed a correlation across studies between amyloid reduction and cognitive outcomes, there is no such correlation within studies, as shown in the analysis released to the public on aducanumab by the FDA Clinical Pharmacology and Biopharmaceutics Review (https://www.fda.gov/media/143504/download; slide 20). Only one study has conducted the same analysis plotting the changes in CDR-SB against ARIA-E. This is a point of concern because trials with more ARIA-E are associated with better outcomes (Fig. 1) [6]. When a patient develops ARIA, the dosing is halted and the patient is asked to undergo more frequent MRI scans until the ARIA resolves. This protocol change almost certainly alerts the patient, their informant, their physician, and other study personnel that the subject is in the active treatment arm of the study. Given that the CDR-SB has an important subjective component, including an interview with the patient’s caregiver, unblinding bias is plausible and may explain a substantial part of the apparent efficacy reported in the trials [9].
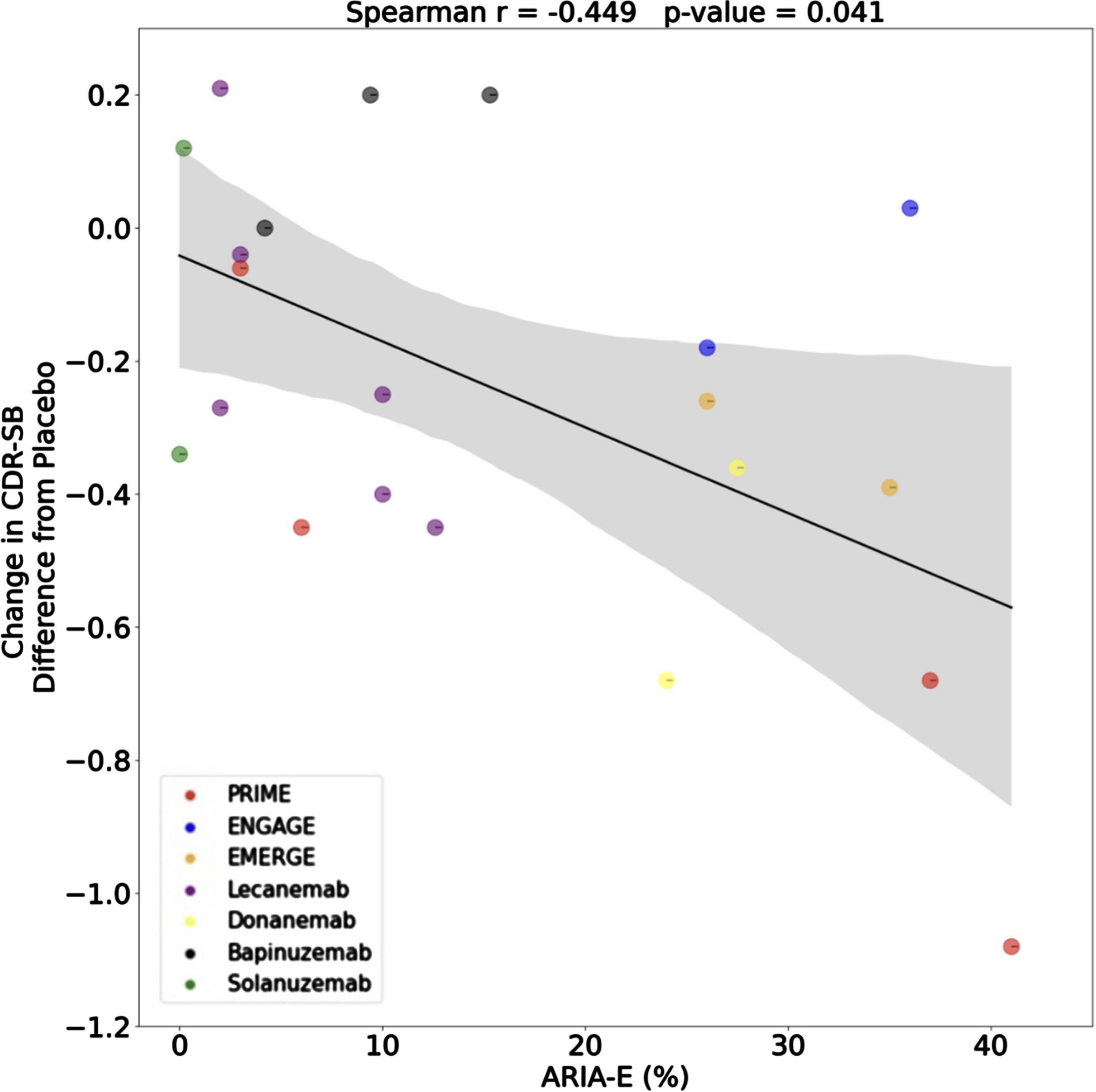
Furthermore, those allocated to the placebo arms received a (non-reactive) saline placebo which cannot mimic the ARIA-generating pharmacological potency of mAbs. A proper control comparison, as done in other trials of highly reactive drugs [10] would attenuate the artificial inflation derived from unblinding, by using two control arms: an active control (e.g., an IgG antibody of similar reactivity, from the same organism, similarly humanized), and a saline placebo, which would carry the largest risk of unblinding, but would come with a smaller risk to the participants [11].
ARIA is caused by local amyloid removal, such that ARIA cannot be separated from efficacy. However, regardless of the exact mechanism of ARIA, unblinding is expected following the general immunogenic reaction and inflammation [12]. Indeed, recent estimates suggest that reactive drug unblinding may explain a substantial part of the apparent efficacy [13].
AS AMYLOID DECREASES, MONOMERIC Aβ42 INCREASES
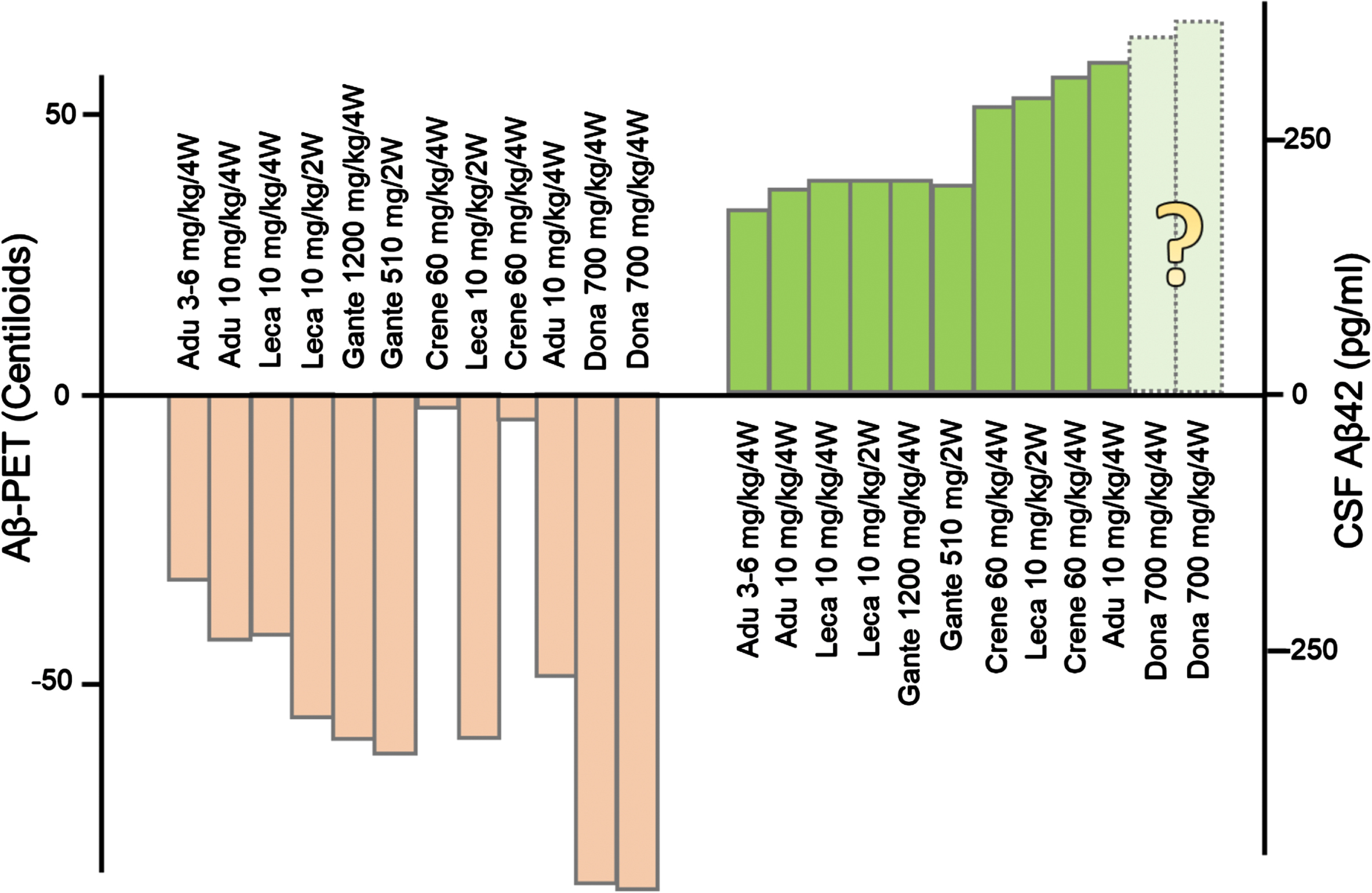
To understand the discrepancies between the large brain Aβ burden reduction and the minimal cognitive benefit, information about the other end of the protein homeostasis is needed. Aducanumab, lecanemab, and other monoclonal antibodies increase the levels of soluble, monomeric Aβ42 in CSF (Fig. 2). We do not know the effects of donanemab on CSF Aβ42 levels because no CSF samples were collected in the TRAILBLAZER-ALZ 2 study. This is an important missing piece of information as it has become increasingly recognized that high brain amyloid is compatible with normal neurological function whereas low soluble Aβ42 is not [14]. The universal increase in CSF Aβ levels induced by monoclonal antibodies [15] may be at least as relevant as the reduction in the insoluble fraction of brain amyloid in determining the cognitive and clinical effects of anti-Aβ monoclonal antibodies. In support of this statement, a recent study found very low levels of CSF Aβ42 (143 pg/mL) in a severe case of autosomal dominant AD due to a rare amyloid precursor protein (APP) mutation (D678N) despite undetectable brain amyloid by PET [16]. The data from the Phase 3 Clarity AD study on lecanemab [1] suggests that the increase in CSF Aβ42 levels could be due to the clearance of brain plaques. The data at 12 months suggest that the marked CSF Aβ42 increase in the initial phase of extensive clearance parallels the marked reduction of Aβ plaques ([1], Figure S5, Supplementary data). Any benefit from the increases in CSF Aβ42 are likely to be short-lived: in the lecanemab trial, the increase in CSF Aβ42 leveled off in the last part of the trial (see [1], Figure S5).
CONCLUDING REMARKS
The most apparently efficacious mAbs all have effects of similar size (2–3% of the absolute primary endpoint scale), all used saline as placebo, and all had similarly large (10–20%) incidences of adverse events, including ARIA. The strong correlation between ARIA and cognitive changes on the more subjective scale but not on more objective scales strongly suggests that functional unblinding by subjects and investigators may have accounted for the differential effect favoring highly-reactive monoclonal antibodies over placebo. In contrast, all antibodies with lower reactivity were ineffective [17]. The unblinding bias is a major issue as it inflates apparent efficacy and thus artificially skews the risk-benefit balance. It is, therefore, essential to recalibrate the nature of the effects of the mAbs and to diversify therapeutic avenues for AD away from those based on removing amyloid.
AUTHOR CONTRIBUTIONS
Alberto J. Espay (Conceptualization; Validation; Writing – original draft; Writing – review & editing); Karl Herrup (Conceptualization; Validation; Writing – original draft; Writing – review & editing); Bruno P. Imbimbo (Conceptualization; Validation; Writing – original draft; Writing – review & editing); Kasper Kepp (Conceptualization; Validation; Writing – original draft; Writing – review & editing); Timothy Daly (Conceptualization; Validation; Writing – original draft; Writing – review & editing).
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
Alberto J. Espay has received grant support from the NIH and the Michael J Fox Foundation; personal compensation as a consultant/scientific advisory board member for Neuroderm, Amneal, Acadia, Avion Pharmaceuticals, Acorda, Kyowa Kirin, Sunovion, Supernus (formerly, USWorldMeds), and Herantis Pharma; personal honoraria for speakership for Avion, Amneal, and Supernus; and publishing royalties from Lippincott Williams & Wilkins, Cambridge University Press, and Springer. He cofounded REGAIN Therapeutics and is co-inventor of the patent “Compositions and methods for treatment and/or prophylaxis of proteinopathies.”
Karl Herrup has no conflicts of interest to report.
Bruno P. Imbimbo is an employee at Chiesi Farmaceutici. He has no stock options. He is co-author of several patents in the field of Alzheimer’s disease.
Kasper P. Kepp has no conflicts of interest to report.
Timothy Daly is an Editorial Board Member of this journal but was not involved in the peer-review process of this article nor had access to any information regarding its peer-review. He has no other conflicts of interest to report.