
Abstract
Background:
Bridging integrator 1 (BIN1) gene polymorphism has been reported to play a role in the pathological processes of Alzheimer’s disease (AD).
Objective:
To explore the association of BIN1 loci with neuroinflammation and AD pathology.
Methods:
Alzheimer’s Disease Neuroimaging Initiative (ADNI, N = 495) was the discovery cohort, and Chinese Alzheimer’s Biomarker and LifestylE (CABLE, N = 619) study was used to replicate the results. Two BIN1 gene polymorphism (rs7561528 and rs744373) were included in the analysis. Multiple linear regression model and causal mediation analysis conducted through 10,000 bootstrapped iterations were used to examine the BIN1 loci relationship with cerebrospinal fluid (CSF) AD biomarkers and alternative biomarker of microglial activation microglia-soluble triggering receptor expressed on myeloid cells 2 (sTREM2).
Results:
In ADNI database, we found a significant association between BIN1 loci (rs7561528 and rs744373) and levels of CSF phosphorylated-tau (P-tau) (pc = 0.017; 0.010, respectively) and total-tau (T-tau) (pc = 0.011; 0.013, respectively). The BIN1 loci were also correlated with CSF sTREM2 levels (pc = 0.010; 0.008, respectively). Mediation analysis demonstrated that CSF sTREM2 partially mediated the association of BIN1 loci with P-tau (Proportion of rs7561528 : 20.8%; Proportion of rs744373 : 24.8%) and T-tau (Proportion of rs7561528 : 36.5%; Proportion of rs744373 : 43.9%). The analysis in CABLE study replicated the mediation role of rs7561528.
Conclusions:
This study demonstrated the correlation between BIN1 loci and CSF AD biomarkers as well as microglia biomarkers. Additionally, the link between BIN1 loci and tau pathology was partially mediated by CSF sTREM2.
INTRODUCTION
The global incidence of dementia is projected to experience a nearly triple surge by the year 2050. 1 Alzheimer’s disease (AD) stands as the predominant form of dementia, specifically late-onset AD (LOAD).2,3, 2,3 The risk of AD is 60–80% dependent on heritable factors, 1 and the Bridging Integrator 1 (BIN1) gene has been reported to be one of the most significant risk factors for LOAD. 4 The influence of BIN1 on AD susceptibility has been consistently observed in Caucasians, 5 as well as within African American populations.6,7, 6,7 Likewise, investigations in East Asian and Han Chinese populations have suggested the impact of BIN1 gene polymorphisms on AD risk.8 –10 Through extensive genome-wide association studies (GWASs) and comprehensive meta-analyses, the most significant connections with AD susceptibility have been established for two single nucleotide polymorphisms (SNPs) within the BIN1 gene, namely rs7561528 and rs744373.6,11,12 , 6,11,12 These SNPs are positioned at distances of about 30 kb and 25 kb from the coding region of BIN1. 13
The primary pathological hallmark of AD centers around the buildup of amyloid-β (Aβ), and the hyperphosphorylation of tau, leading to the development of intracellular neurofibrillary tangles (NFTs). BIN1 is likely involved in AD as a modulator of NFT pathology, and this role may extend to tau pathology. 14 Other studies also suggested that BIN1 mediated AD risk by modulating tau pathology.13,15, 13,15 The association of BIN1 loci with tau pathology was also confirmed in tau-PET. 16 Previous studies have shown that BIN1 expression has been observed to be heightened in AD brains, particularly within neurons containing NFTs. 17 Interestingly, recent studies have discovered pronounced expression of BIN1 mainly within microglial cells. 18 Microglia play a critical role in orchestrating neuroinflammatory processes, exerting their influence on brain aging, and the progression of neurodegenerative conditions. 19 The microglial isotype of the BIN1 protein was increased while the neuronal isotype was decreased. BIN1 variations and abnormal expression are mainly related to microglia in the brain, but not to neurons and astrocytes. 18 Notably, the activation of microglia, prompted by microglia-soluble triggering receptor expressed on myeloid cells 2 (sTREM2), elicits inflammatory responses that contribute to both protective and survival-promoting effects. 20 The presence of sTREM2 in cerebrospinal fluid (CSF) has been suggested as an alternative biomarker of microglial activation in many neurodegenerative diseases. 21 Besides, neuroinflammation is associated with the progression of tau pathology.22,23, 22,23 These results provide some clues for a potential relationship between BIN1 and microglial activation in AD pathogenesis.
Therefore, we aimed to analyze the associations of BIN1 two loci (rs7561528 and rs744373) with CSF AD biomarkers and microglial biomarker sTREM2 levels; as well as to evaluate whether the correlation between BIN1 loci and AD pathology is influenced by CSF sTREM2. To achieve these goals, we analyzed data retrieved from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort and replicated the results in the Chinese Alzheimer’s Biomarker and LifestylE (CABLE) study.
METHODS
ADNI dataset
Our analysis included 38 diagnosed with AD, 292 with mild cognitive impairment (MCI), and 165 with cognitive normal (CN) from the ADNI database (http://adni.loni.usc.edu/), which was funded as a private-public partnership, ADNI began in 2004 and is overseen by Michael W. Weiner, MD, of the VA Medical Center and the University of California-San Francisco. All participants provided written informed consent according to the Declaration of Helsinki before study enrollment. The institutional review boards of all participating institutions in ADNI approved the data used for this study. To mitigate the risk of spurious results stemming from potential population stratification effects, we excluded a total of 50 individuals belonging to the non-Hispanic white group. Considering the use of Aβ42 for grouping, cases that are close to the cutoff value are prone to be misclassified due to measurement errors in the variable. Hence, to ensure that our conclusions are based on a consistent and robust framework, we opted to exclude any data falling within the confidence interval of±5% around the initial cutoff value, a methodology that has been employed in prior research.24 –27 Consequently, we removed 199 individuals from our analysis due to their association with gray zones or having missing Aβ42 values. Ultimately, our analysis was conducted on a cohort of 495 individuals at the baseline, all of whom exhibited AD core biomarkers (Supplementary Figure 1).
SNPs selection
The BIN1 genotypes were derived from the ADNI dataset in PLINK format. 28 To date, robust associations between BIN1 loci (specifically rs7561528 and rs744373) and AD have been consistently identified in previous genome-wide association studies.4,11,15,29 , 4,11,15,29 We selected two representative sites (rs7561528 and rs744373) as BIN1 sites for this study. Next, we executed quality control (QC) procedures employing the PLINK software. Our inclusion criteria encompassed the following thresholds: a minimum call rate exceeding 90%, minimum minor allele frequencies (MAF) greater than 0.01, and a p-value greater than 0.001 in the Hardy-Weinberg equilibrium test.
CSF biomarker data
CSF levels of key AD biomarkers, including Aβ42, phosphorylated-tau (P-tau), and total-tau (T-tau), were evaluated through the employment of the multiplex xMAP Luminex platform, developed by Luminex Corp in Austin, TX. This assessment was conducted using Innogenetics’ immunoassay kit-based reagents (INNO-BIA AlzBio3) from Ghent, Belgium, specifically intended for research purposes. These measurements were performed at the ADNI Biomarker Core Laboratory situated at the University of Pennsylvania. To ensure accuracy, duplicate measurements were taken and subsequently averaged. Additionally, CSF sTREM2 levels were quantified using the MSD platform-based assay, a method that has been previously published and validated. 30
Grouping of subjects
Initially, the study participants were categorized according to their cognitive diagnoses, resulting in 38 individuals with AD, 292 with MCI, and 165 classified as CN. Subsequently, they were further divided into two groups based on their Aβ status: those with abnormal Aβ levels (A+) and those with normal Aβ levels (A–). This division was determined using baseline CSF measurements with predefined cutoffs for CSF Aβ1 - 42 (192 ng/L), as specified in the ADNI database. 31 Furthermore, participants were stratified based on their apolipoprotein E (APOE) ɛ4 allele status, distinguishing between carriers and non-carriers. This separation aimed to investigate the potential influences of the APOE ɛ4 allele on the relationship between BIN1 loci and the levels of AD biomarkers as well as sTREM2.
CABLE study
To validate the findings from ADNI, data of 619 adults without dementia were downloaded from the CABLE study. The CABLE study is an ongoing large and independent cohort focused on determining the genetic and environmental modifiers of AD biomarkers and their utility in the early diagnosis in the northern Chinese Han population.32,33, 32,33 The Institutional Ethics Committee of Qingdao Municipal Hospital approved the CABLE study, and it was carried out following the Declaration of Helsinki. All subjects or their proxies gave written consent. All participants had complete information including age, gender, years of education, APOE ɛ4 status, two BIN1 SNPs (rs7561528, rs744373) expression data, and levels of CSF AD core biomarkers including Aβ42, P-tau, T-tau, and sTREM2. Detailed quality control information is shown in Supplementary Methods. The ADNI database includes many subjects with MCI, but most participants in the CABLE study were cognitively normal, reflecting early disease changes. And the proportion of the A + population (23%) was not high. Therefore, we did not identify interaction or subgroup effects stratified by Aβ burden in the CABLE study.
Statistical analyses
Our statistical analyses were conducted using R 4.2.1 (http://www.r-project.org/) and PLINK 1.07 (http://pngu.mgh.harvard.edu/wpurcell/plink/). BIN1 genotypes were classified according to the presence or absence of the minor allele. In this study, APOE ɛ4 status was grouped into individuals carrying at least one APOE ɛ4 allele or individuals with no APOE ɛ4 allele. To explore differences in CSF biomarkers and sociodemographic data, t-tests were used for continuous variables with a normal distribution, and the Mann–Whitney U test was used for continuous variables that did not follow a normal distribution. Chi-square tests were employed for dichotomous variables such as APOE ɛ4 status and gender. The CSF biomarker levels did not follow a normal distribution (Kolmogorov–Smirnov test: p < 0.01). Therefore, the levels were log10 transformed to approach a normal distribution for the following analysis. After adjusting for potential confounding factors, we used the one-way analysis of covariance (ANCOVA) for further comparisons, followed by Fisher’s LSD as a post hoc test. We employed a multiple linear regression model, including age, gender, educational level, APOE ɛ4 status, and diagnosis as covariates, to estimate possible associations between the allele carrier status of the two BIN1 SNPs (rs7561528 and rs744373) and AD-related CSF biomarkers at baseline among ADNI participants. To control for type I errors arising from multiple tests, we applied the false discovery rate (FDR) method developed by Hochberg and Benjamini. 34 We considered a significance threshold of pc < 0.05 following FDR correction.
Our primary hypothesis centered on utilizing CSF sTREM2 as a potential mediator, to assess whether the links between BIN1 loci and AD pathologies could be mediated through CSF sTREM2. To investigate this, we employed mediation analyses based on the method developed by Baron and Kenny. 35 The presence of mediation effects was considered if the following criteria were simultaneously met: (1) a significant association between BIN1 and CSF AD biomarkers (P-tau, T-tau, P-tau/Aβ42, Tau/Aβ42); (2) a significant association between BIN1 and CSF sTREM2; (3) a significant association between CSF AD biomarkers and CSF sTREM2; and (4) the inclusion of CSF sTREM2 as a mediator led to the reduction of the associations between BIN1 loci and CSF AD biomarkers. Additionally, we determined significance through 10,000 bootstrapped iterations using the “mediate” package in R 4.2.1 software.
RESULTS
Participant characteristics
Table 1 provides a summary of the demographic details for the included subjects. Among these subjects, 283 carried the A allele (AG+AA) of rs7561528, while 243 were carriers of the C allele (TC+CC) of rs744373 in the ADNI dataset. Notably, there were significant differences observed in the levels of CSF biomarkers, including P-tau, T-tau, P-tau/Aβ42, T-tau/Aβ42, and sTREM2, between BIN1 allele carriers and non-carriers (p < 0.05). However, there were no significant differences found in age, gender distribution, or education levels between these two groups (p > 0.05). Furthermore, Table 1 also presents the characteristics of participants from the CABLE study.
Characteristics of participants in ADNI and CABLE
p-value represents the difference between two groups, significant effects (p < 0.05) are shown in bold. Categorical variables were reported as numbers; continuous variables were reported as means (SDs). Intergroup differences were assessed using chi-square analysis for categorical data, t-tests for data with a normal distribution, and the Mann–Whitney U test for data that did not follow a normal distribution. CSF, cerebrospinal fluid; Aβ42, amyloid-β42; P-tau, phosphorylated tau protein; T-tau, total tau protein; sTREM2, soluble triggering receptor expressed on myeloid cells 2; SD, standard deviation.
Differences in AD biomarkers between BIN1 genotype allele states in the ADNI database
In the initial phase, we employed a one-way ANCOVA to assess whether there were disparities in the levels of Aβ42, P-tau, T-tau, P-tau/Aβ42, Tau/Aβ42, and sTREM2 between BIN1 allele carriers and non-carriers. In the ADNI dataset, we observed that Aβ42 did not exhibit any discernible difference between BIN1 alleles. However, we did observe significant distinctions in tau pathology between individuals with A genotypes (AG+AA) and GG genotypes at rs7561528 (P-tau: p = 0.015; T-tau: p = 0.003; P-tau/Aβ42: p = 0.016; T-tau/Aβ42: p = 0.003), as well as between individuals with C-carrying genotypes (TC+CC) and TT genotypes at rs744373 ((P-tau: p = 0.011; T-tau: p = 0.002; P-tau/Aβ42: p = 0.023; T-tau/Aβ42: p = 0.005) (Fig. 1; Supplementary Figure 2).
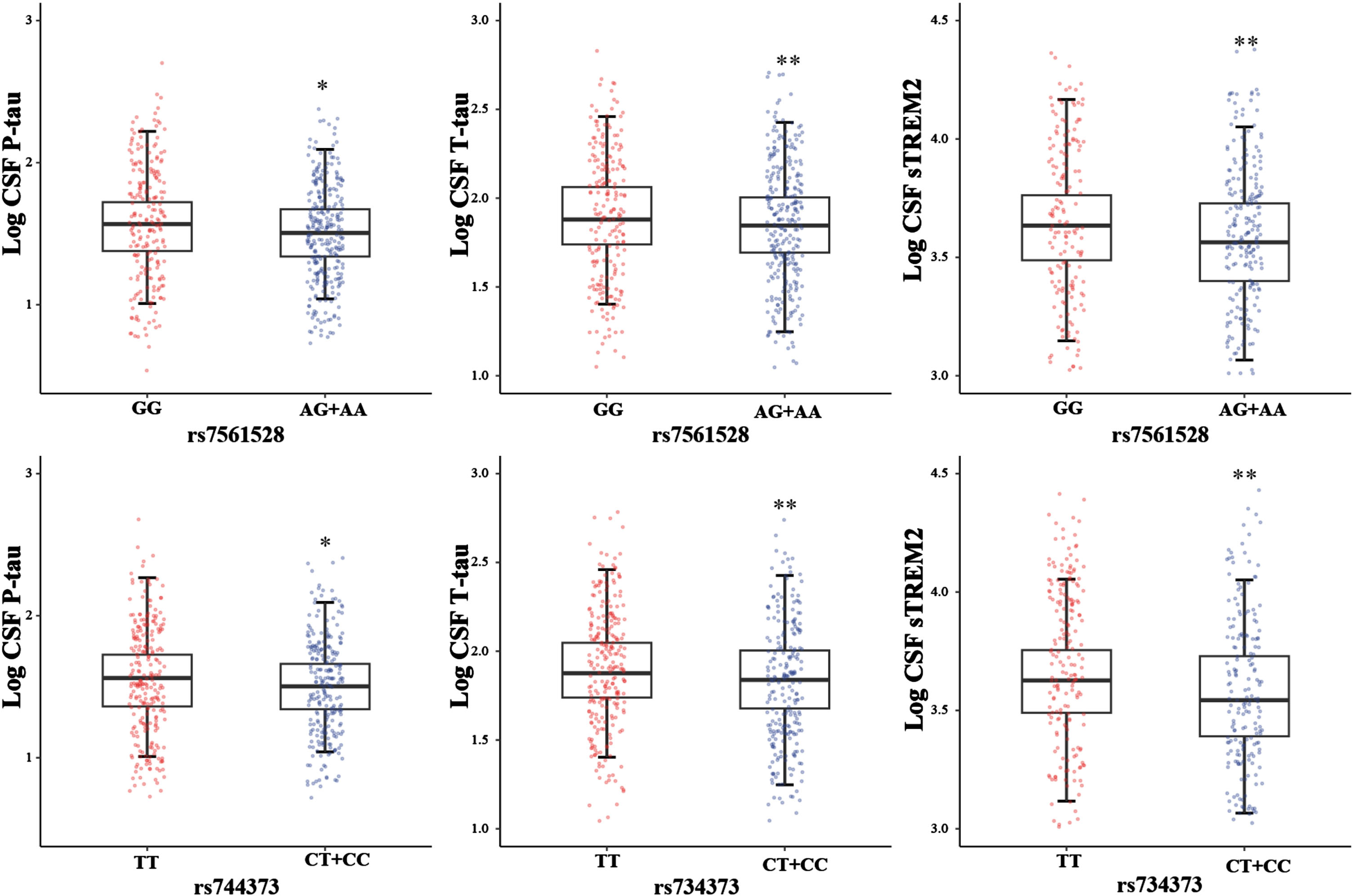
Association of BIN1 loci with CSF AD biomarkers in the ADNI database
Multiple linear regression analyses were conducted within the ADNI sample, with adjustments made for age, gender, educational level, APOE ɛ4 status, and diagnosis. Minor allele carriers of BIN1 rs7565128 exhibited associations with lower CSF P-tau levels (β = -0.048, pc = 0.017), T-tau levels (β = -0.051, pc = 0.011), P-tau/Aβ42 (β = -0.060, pc = 0.019), T-tau/Aβ42 (β = -0.064, pc = 0.011), and sTREM2 (β = -0.065, pc = 0.010). However, there was no association with CSF Aβ42 levels in all participants, indicating a robust connection between BIN1 and brain tau pathology. Similarly, no significant relationship was found between Aβ42 levels and rs744373. For minor allele carriers of BIN1 rs744373, there were associations with lower CSF P-tau levels (β = -0.053, pc = 0.010), CSF T-tau levels (β = -0.058, pc = 0.013), P-tau/Aβ42 (β = -0.064, pc = 0.014), T-tau/Aβ42 (β = -0.071, pc = 0.008), and sTREM2 (β = -0.070, pc = 0.008) as determined by the FDR test. Notably, similar and more significant associations were observed in the A + subgroup (rs744373: T-tau p = 0.004, P-tau p = 0.005, and sTREM2 p = 0.005; rs7561528: T-tau p = 0.005, P-tau p = 0.004, and sTREM2 p = 0.013). Subgroup analyses revealed that these findings also held in APOE ɛ4 carrier subgroup (rs744373: T-tau p = 0.007, P-tau p = 0.013, and sTREM2 p = 0.009; rs7561528: T-tau p = 0.011, P-tau p = 0.023, and sTREM2 p = 0.012), as well as in the MCI subgroups, but not in A-, CN, and APOE ɛ4 non-carrier subgroups (p > 0.05) (Fig. 2; Supplementary Table 1). In our longitudinal analysis of the ADNI database, no statistical evidence emerged for an effect of the minor allele carriers of rs7561528 and rs744373 in BIN1 on longitudinal CSF biomarkers in the total group, even after accounting for age, gender, educational level, APOE ɛ4 status, and diagnosis (Supplementary Table 2).
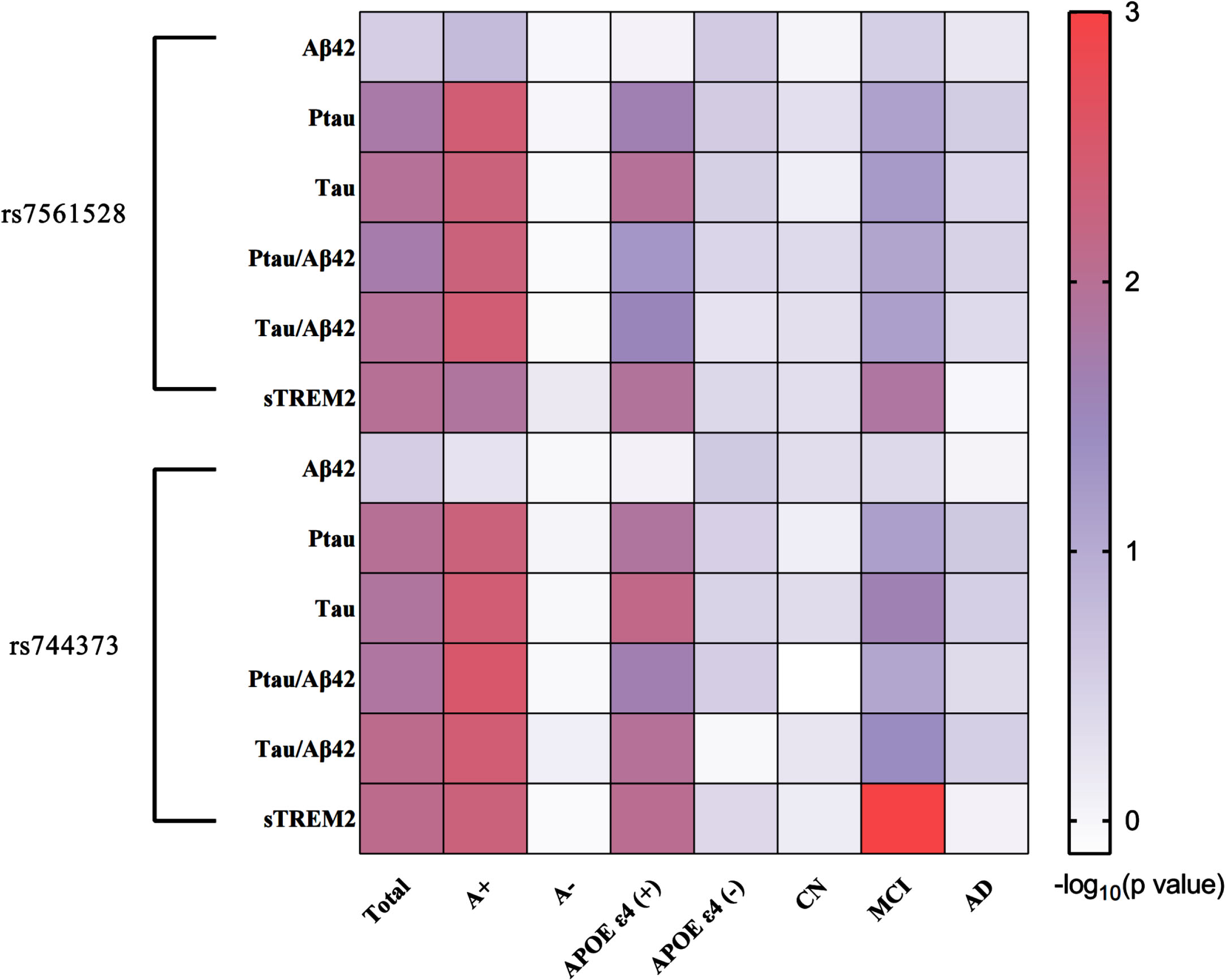
Mediation analyses in the ADNI database
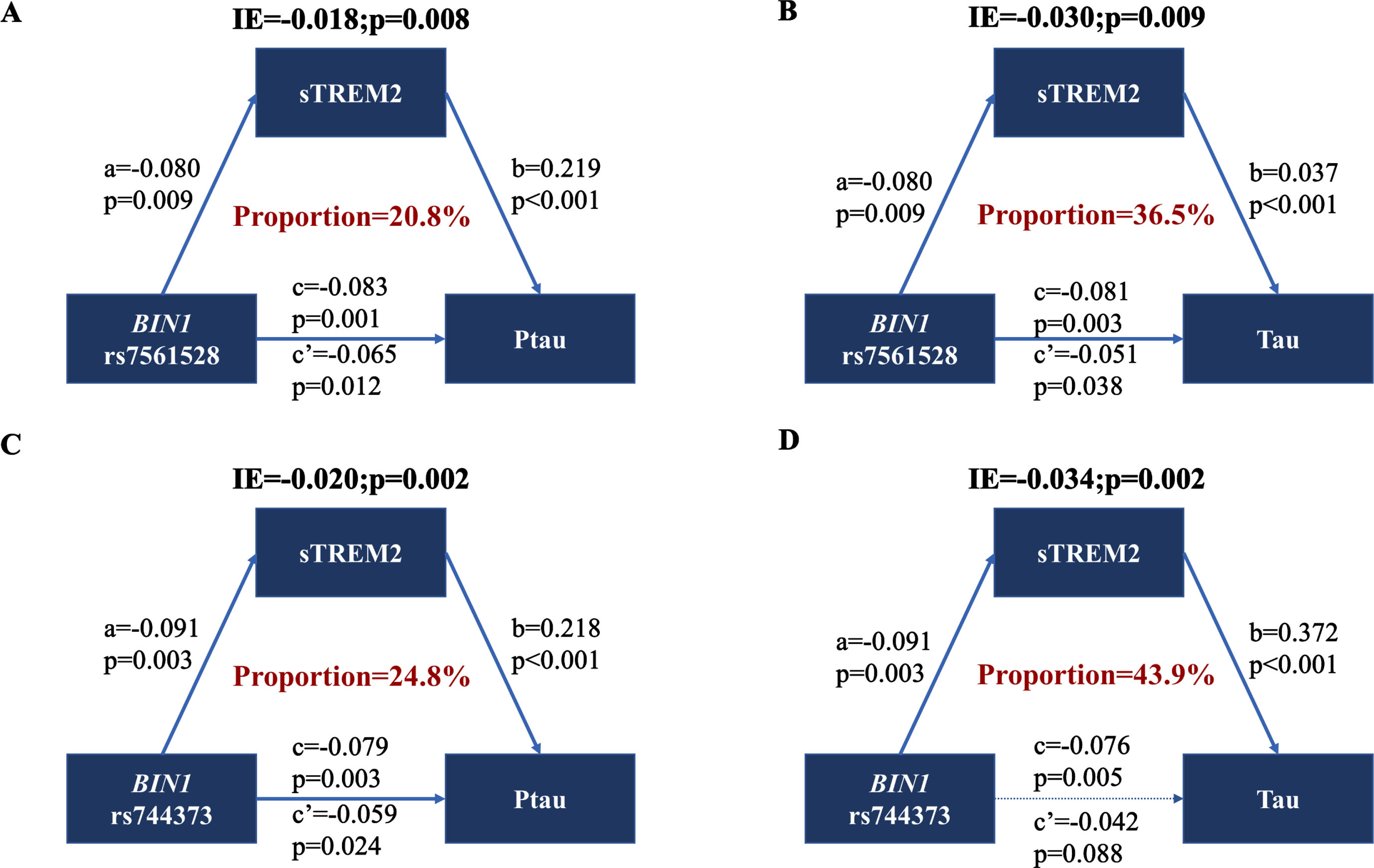
Building upon the previous findings, our next step was to examine the primary hypothesis concerning the potential involvement of CSF sTREM2 in the relationship between BIN1 loci and AD pathology. To investigate this, we conducted mediation analyses for CSF P-tau, T-tau, P-tau/Aβ42, and T-tau/Aβ42 (Supplementary Table 3). Results of mediation analyses showed that the association of the minor allele carriers of rs7561528 and rs744373 in BIN1 and AD-related biomarkers was partially mediated by CSF sTREM2 in the A + subgroup (Fig. 3; Supplementary Figure 3). The proportion of mediation ranged from 14.8% to 43.9%.
Replication in CABLE study
We conducted a replication analysis using an independent cohort comprising 619 participants from the CABLE study. The outcomes derived from the one-way ANCOVA analysis unveiled notable distinctions in tau pathology and sTREM2 levels between individuals carrying the BIN1 rs7561528 allele and those who did not, within the confines of this CABLE study cohort (P-tau: p = 0.003; T-tau: p = 0.015; sTREM2: p = 0.020) (Fig. 4A-C). However, there was no significant association found between the BIN1 rs744373 allele state and AD pathology in this particular cohort.

In our analysis, we utilized a linear regression model to investigate the connection between BIN1 and tau pathology. This examination was carried out with due consideration for factors such as age, gender, educational level, and APOE ɛ4 status. The results indicated that individuals with BIN1 rs7561528 AA and GA genotypes exhibited lower levels of CSF P-tau (β = -0.033, p = 0.003), T-tau (β = -0.045, p = 0.015), and sTREM2 (β = -0.049, p = 0.020) when compared to the GG group. However, BIN1 loci at rs7565128 did not show any significant association with the level of CSF Aβ42 (β = -0.028, p = 0.186), which aligns with the findings from the ADNI database. On the other hand, there was no significant association detected between BIN1 loci at rs744373 and AD core biomarkers within the CABLE study.
The mediation effects were successfully replicated in the CABLE study. Similarly, we observed that the association between BIN1 rs7561528 and tau pathology was partially mediated by CSF sTREM2. The extent of mediation ranged from 23.2% to 29.1% (p < 0.05) (Fig. 4D, E; Supplementary Table 4). Furthermore, the indirect and total effects of CSF sTREM2 at rs744373 on CSF Tau (Fig. 4E) were statistically significant (p < 0.05), while the direct effect did not reach statistical significance (p > 0.05), which could be related to the masking effect. 36
DISCUSSION
This is a large-scale study to assess the interrelationships between BIN1 loci (rs7561528 and rs744373), CSF sTREM2, and AD-related biomarkers. We indicated that BIN1 loci (rs7561528 and rs744373) are significantly related to CSF sTREM2 and tau pathology. Mediation analysis results showed that sTREM2-mediated BIN1 gene polymorphism was associated with tau pathology. Especially, BIN1 rs7561528 was verified in two independent cohorts.
Our results are consistent with previously reported studies showing no significant association between BIN1 loci and CSF levels of Aβ. 37 but it was associated with changes in CSF P-tau and T-tau levels. 38 Carriers of the minor allele of BIN1 rs744373 exhibited lower CSF P-tau levels. 39 Furthermore, our analysis revealed associations between BIN1 rs744373 and rs7561528 with P-tau/Aβ42 and T-tau/Aβ42 in the ADNI dataset. Subsequently, when stratified the Knight ADRC and the MDC Brain Bank by BIN1 (rs7561528) genotypes, a significant impact of the BIN1 gene’s G allele on TREM2 expression was observed. Specifically, homozygous GG carriers displayed higher TREM2 levels compared to heterozygous AG carriers. 40 Similarly, we found that BIN1 allele carriers had lower CSF sTREM2 levels. As far as our current understanding goes, we have presented the inaugural report indicating that CSF sTREM2 could serve as a partial mediator in conveying the effects of BIN1 polymorphism on tau pathology.
Whether BIN1 causes tau pathological changes through neuroinflammation-related pathways is not completely clear, but some previous studies have given some hints. Many large GWAS have identified BIN1 as strongly associated with AD. Previous studies identified BIN1 is over-expressed in the brain of AD cases. 15 Recent research has revealed a significant development: the removal of a microglia-specific enhancer containing AD-risk variants led to the absence of BIN1 expression specifically within microglia, while its expression in neurons or astrocytes remained unaltered. 18 This discovery underscores that the most substantial risk allele identified in GWAS associated with BIN1 resides within a microglia-specific enhancer region. BIN1 is a member of the BIN1/Amphiphysin/RVS167 (BAR) family of adaptor proteins, which are integral in regulating the dynamics of lipid membranes.41,42, 41,42 This multifaceted protein has been implicated in a wide range of cellular processes, including endocytosis, actin dynamics, DNA repair, membrane trafficking, inflammation, and apoptosis.42,43, 42,43 Significantly, BIN1 has also been linked to the process of phagocytosis in macrophages. 44 Microglia, on the other hand, are tissue-resident macrophages situated within the central nervous system (CNS). They play a pivotal role in immune defense, CNS development, and maintaining homeostasis. 45 BIN1 expression plays a central role in regulating microglial responses to challenges in the CNS, and it’s suggested that the risk of AD might stem from impaired BIN1 function within microglia. 46 Microglia function as a double-edged sword in AD. As a specific protein of microglia, TREM2 has been reported to allow microglia to counteract the detrimental Aβ plaque deposition, then promoting memory function. 47 The role of TREM2 in tau pathology appears contradictory between the early and late stages of AD. 48 TREM2-activating antibodies can enhance microglial migration towards amyloid plaques, promote phagocytosis, reduce endogenous tau hyperphosphorylation, and improve cognitive function. 49 On the other hand, if TREM2 persistently activates microglia without robust amyloid clearance, may exacerbate Aβ-induced tau pathology. 50 Additionally, elevated levels of sTREM2 have been detected in the cerebrospinal fluid of patients in the early stages of AD. These elevated sTREM2 levels are positively correlated with classic cerebrospinal fluid markers, total tau, and phosphorylated tau, but they do not show a correlation with Aβ, APOE4 status, or gender. 51 Furthermore, there is mounting evidence indicating that sTREM2 might also participate in microglial dynamics and their response to AD-related pathology, especially tau pathology. 51 In vitro experiments suggest that sTREM2 can enhance the viability of microglial cells and promote inflammatory responses by activating signaling pathways such as Akt–GSK3β–β-catenin and NF-κB. 20 In this study, the results indicated that CSF sTREM2 could mediate in conveying the effects of BIN1 polymorphism on tau pathology, providing a new possibility for the pathological mechanism of microglia and tau pathology.
SNPs in the BIN1 gene that are associated with AD show a significant correlation with tau pathology while displaying no notable connection with Aβ levels in the CSF and brains of individuals affected by AD.15,38, 15,38 BIN1 expression in forebrain neurons promoted neuroinflammation and tau pathogenesis in the hippocampus, and deletion of BIN1 also attenuated region-specific tau neuropathology, thereby attenuating neuronal death and brain atrophy. 52 Microglial BIN1 was found to influence the release of tau in extracellular vesicles. 53 Collectively, the evidence presented, including our findings, suggests that BIN1 polymorphisms may influence tau protein expression (markers of neuronal degeneration/injury) through sTREM2, thereby mediating the risk of AD. However, the precise biological mechanisms underpinning this association warrant further exploration in future research endeavors.
Interestingly, subgroup analyses further showed that associations were more meaningful in the A + subgroup, while no significance was observed in the A–subgroup. The prevailing preclinical AD model suggests that Aβ pathology serves as the initial trigger, followed by subsequent neurodegenerative alterations. This perspective reinforces the concept that the presence of Aβ contributes to the augmentation of tau pathology. 54 It was also reported that the progression of tau pathology in AD may necessitate Aβ deposition. 55 A study using the ADNI and BioFINDER cohort found that BIN1 risk-allele carriers exhibited accelerated tau pathological changes at more abnormal Aβ levels. 16 The role of TREM2 may depend on Aβ pathology and the stage of the disease. 48 Existing studies have also shown that cerebrospinal fluid sTREM2 levels decrease only in the presence of abnormal Aβ pathology in the early asymptomatic stage of AD. 56 The second interesting observation is that among the two BIN1 loci, only one BIN1 locus, rs7561528, has been validated in the Asian population, and no corresponding results have been observed for the other locus. This suggests that there may be some racial differences. Therefore, considering potential racial differences when investigating the underlying mechanisms of BIN1 becomes an important topic for future discussion.
The strengths of the current study are as follows: Firstly, our study demonstrated that two sites of BIN1 are significantly associated with tau pathology. To the best of our knowledge, we report for the first time that CSF sTREM2 partially mediates the relationship between these gene loci and tau pathology. Furthermore, this finding provided a new possible avenue for targeting inflammatory pathways to impact tau pathology through early interventions which may potentially mitigate the development of tau pathology in the future. Secondly, our study benefited from the inclusion of two separate cohorts, enabling us to replicate our findings in one cohort with another. The ADNI database participants included in this study were white and the CABLE study participants were all Han Chinese, confidence should be exercised in generalizing these findings to other ethnic groups. Finally, the cohort in our study comprised individuals with mainly non-AD, symbolizing the early disease-related changes that carry significant implications for disease prevention and progression. Future research could utilize BIN1 variant loci for screening high-risk populations, which would facilitate early detection of genetic influences on cognitive function. Nonetheless, it is crucial to recognize the constraints of our present study. First of all, the longitudinal follow-up data are lacking, resulting in limiting exploration of causal mechanisms. Secondly, the impact of other genetic mutations on the results also needs to be considered, such as the TREM2 mutation. 57 Besides, follow-up measurements should be studied to validate our findings in larger and longitudinal cohorts to get a robust conclusion with causality. In addition, more biomarkers like concentration of BIN1 protein, neuroinflammatory factors, microglia, astrocytes, and synaptic markers could help to explore their changes and potential mechanisms in different BIN1 mutations and stages of AD. Subsequently, more cellular and animal experiments are needed to further explore the detailed mechanisms of the present finding.
Conclusion
In summary, our study has discovered a notable correlation between BIN1 loci (rs7561528 and rs744373) and the levels of tau protein and CSF sTREM2. CSF sTREM2 acts as an intermediary, connecting BIN1 gene polymorphisms to tau pathology, especially in populations with positive Aβ deposition. However, further studies utilizing cellular and animal models are necessary to determine the specific mechanism by which genetic variations in BIN1 impact tau pathology.
AUTHORS CONTRIBUTIONS
Fan Guo (Conceptualization; Formal analysis; Investigation; Methodology; Validation; Writing – original draft); Meng-Shan Tan (Funding acquisition; Resources; Supervision; Validation; Writing – review & editing); Hu Hao (Data curation; Funding acquisition; Resources; Validation; Visualization); Ya-Nan Ou (Conceptualization; Project administration; Supervision; Writing – review & editing); Ming-Zhan Zhang (Formal analysis; Investigation; Software); Ze-Hu Sheng (Data curation; Methodology; Software); Hao-Chen Chi (Investigation; Software); Lan Tan (Funding acquisition; Project administration; Resources; Supervision; Writing – review & editing).
Footnotes
ACKNOWLEDGMENTS
Data collection and sharing for this project was funded by ADNI (National Institutes of Health Grant U01 AG024904) and (W81XWH-12-2-0012). ![]() .
.
The authors thank the colleagues who have made contributions to build the CABLE cohort. The authors also thank the subjects and their family for their cooperation in this study.
FUNDING
This study was supported by grants from the National Natural Science Foundation of China (82271475 and 82201587), Taishan Scholars Program of Shandong Province (tsqn20161078), Natural Science Foundation of Shandong Province (ZR2023MH062), and Medical Science Research Guidance Plan of Qingdao (2021-WJZD001).
CONFLICT OF INTEREST
Dr. Lan Tan is an Editorial Board Member of this journal but was not involved in the peer-review process of this article nor had access to any information regarding its peer-review.
All other authors have no conflict of interest to report.
DATA AVAILABILITY
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.