
Abstract
This article examines the relationship between cholesterol levels and Alzheimer’s disease (AD), beginning with the early observation that individuals who died from heart attacks often had brain amyloid deposition. Subsequent animal model research proved that high cholesterol could hasten amyloid accumulation. In contrast, cholesterol-lowering treatments appeared to counteract this effect. Human autopsy studies reinforced the cholesterol-AD connection, revealing that higher cholesterol levels during midlife significantly correlated with higher brain amyloid pathology. This effect was especially pronounced in individuals aged 40 to 55. Epidemiological data supported animal research and human tissue observations and suggested that managing cholesterol levels in midlife could reduce the risk of developing AD. We analyze the main observational studies and clinical trials on the efficacy of statins. While observational data often suggest a potential protective effect against AD, clinical trials have not consistently shown benefit. The failure of these trials to demonstrate a clear advantage is partially attributed to multiple factors, including the timing of statin therapy, the type of statin and the appropriate selection of patients for treatment. Many studies failed to target individuals who might benefit most from early intervention, such as high-risk patients like APOE4 carriers. The review addresses how cholesterol is implicated in AD through various biological pathways, the potential preventive role of cholesterol management as suggested by observational studies, and the difficulties encountered in clinical trials, particularly related to statin use. The paper highlights the need to explore alternate therapeutic targets and mechanisms that escape statin intervention.
INTRODUCTION
Over three decades ago, a complex relationship between hypercholesterolemia and Alzheimer’s disease (AD) began to emerge. Larry Sparks noted that patients who succumbed to myocardial infarction often presented with amyloid pathology in their brains, a key neuropathological feature of AD [1]. This observation set the stage for the work of Larry Refolo and co-investigators. Their studies used transgenic mouse models of AD to demonstrate that diet-inducing hypercholesterolemia significantly accelerated amyloid deposition [2]. The investigators showed that when these AD transgenic mice were treated with cholesterol-lowering drugs, there was a marked decrease in amyloid deposition [3]. This research was conducted blindly; mice were treated in Refolo’s laboratory, while the neuropathological evaluation and image analysis were independently performed by one of the authors (MAP), who remained blind to the treatments administered.
Collectively, these investigations highlight a strong mechanistic association between cholesterol and AD pathogenesis, thereby paving the way for further research.
Further evidence of the cholesterol-AD connection emerged from human autopsy studies, revealing a robust correlation between midlife cholesterol levels and subsequent brain amyloid accumulation. This association was particularly pronounced in subjects aged 40 to 55, where even a moderate increase in serum cholesterol, from 181 to 200 mg/dl, nearly tripled the risk of developing brain amyloid, independent of apolipoprotein E (APOE) isoform [4]. Intriguingly, this observation faded with age, pointing toward hypercholesterolemia as being, for unknown reasons, only an early risk factor for AD.
Several epidemiological studies also substantiated the role of midlife hypercholesterolemia in impacting AD risk [5–7]. The first study was conducted by Notkola et al. who investigated the relationship between serum total cholesterol, the APOEɛ4 allele, and AD in a cohort of 444 men aged 70–89 [5]. They found that a previous high serum cholesterol level at mid-life was significantly associated with an increased prevalence of AD later in life, independent of the APOE4 allele’s presence. The research suggests that elevated cholesterol might be an independent risk factor for AD, and the influence of the APOE4 allele on AD risk could be partly mediated through its impact on cholesterol levels. This study supported the concept that managing cholesterol levels in mid-life, before the clinical symptoms of AD manifest, might be crucial in preventing or delaying the onset of AD later in life.
One of these studies examined a multiethnic cohort comprising 9,844 participants who underwent detailed health evaluations at ages 40– 45 [7]. These results revealed that even moderately elevated cholesterol levels were associated with an increased risk of developing late onset AD, reinforcing, as emphasized below in the chapter, the imperative to address dementia risk factors early, perhaps not later than during midlife and decidedly before developing cognitive impairment later in life.
Power and colleagues studied the Atherosclerosis Risk in Communities (ARIC) dataset, which involved nearly 14,000 participants, to understand the long-term impact of midlife cholesterol on cognitive health [8]. They reported that elevated levels of total cholesterol, low-density lipoprotein cholesterol (LDL-c), and triglycerides during midlife were associated with a significant decline in executive function, sustained attention, and processing speed over the ensuing two decades. Additionally, higher total cholesterol and triglycerides were linked with a more marked decline in memory scores. Notably, these investigators showed that high-density lipoprotein cholesterol (HDL-c) did not correlate significantly with cognitive change (a finding refuted by another study discussed below). All these findings emphasized the contribution of hypercholesterolemia as an early risk factor for AD, highlighting the potential benefits of early cholesterol management for long-term harvesting of cognitive health.
While a substantial body of research suggested that high cholesterol levels in mid-life are strongly associated with an increased AD risk later in life, some studies focusing primarily on older populations presented conflicting results. Reitz et al. [9], for example, observed that in individuals aged 77 and older, higher total cholesterol levels paradoxically appeared to decrease the risk of AD (HR = 0.48, 95% CI = 0.26– 0.86), without significant distinctions between HDL and LDL cholesterol. This trend was also evident in their subsequent study, which did not find a significant impact of cholesterol on cognitive function in the elderly [10]. Similarly, another study by Reitz and colleagues [11] indicated that high total cholesterol or LDL levels in those 65 and older were paradoxically correlated with a reduced risk of developing mild cognitive impairment (MCI). Mielke et al. [12] reported that elevated cholesterol levels between ages 70–79 were associated with a lower dementia risk from ages 79–88. These findings contrast with the epidemiological studies that examined younger subjects, which consistently link higher mid-life cholesterol levels to a greater AD risk in later life.
In a review paper published by Sánchez-Ferro and colleagues, the investigators highlighted the significance of the timing of data collection to the disease process when examining the relationship between blood pressure, body mass index (BMI), cholesterol and dementia [13]. The researchers found that studies with less than a decade of follow-up often report no relationship or one that contradicts expected trends between these factors and dementia risk. Conversely, in studies extending beyond ten years of follow-up, arterial hypertension, cholesterol levels, and elevated BMI have consistently been linked to an increased risk of AD. This discrepancy is believed to stem from the natural course of dementia, where cholesterol, blood pressure, and BMI begin to decrease several years before the clinical onset of the disease. Initially, Notkola et al. supported this perspective, noting that gradual decreases in cholesterol levels precede the onset of dementia by several years, thereby potentially masking earlier life hypercholesterolemia. Potential additional factors are discussed in an excellent review by Shepardson et al. [14].
The CRISP Pilot Study evaluated the impact of lovastatin on the health-related quality of life in older individuals, primarily aged 65 or above, focusing on domains like physical functioning, cognitive function, and overall health perception [15]. Despite reduced cholesterol levels with lovastatin treatment, no significant changes in health-related quality of life measures were observed after six months. The negative results could be attributed to the older age of participants and the short follow-up period, which is likely insufficient to observe changes in quality of life or cognitive function in response to lipid-lowering therapy.
The Honolulu-Asia Aging Study by Kalmijn et al. assessed the long-term impact of metabolic cardiovascular syndrome in middle-aged Japanese-American men on their risk of developing dementia in later life [16]. The study, initiated in 1965, followed participants into old age, diagnosing 215 dementia cases. It found that increased metabolic risk factors were associated with a higher risk of vascular dementia but not AD. Again, the relatively advanced age of participants at the onset of the study may not accurately reflect the potential preventive impact of addressing metabolic factors earlier inlife.
MECHANISMS
Mechanistically, other obstacles emerged on the road toward a clear understanding. While Refolo’s transgenic mice data suggested a clear, linear relationship between cholesterol and amyloid load (Fig. 2), this relationship was much more complex in human brain tissue (Fig. 3), stressing the importance of additional modulating factors impacting amyloid deposition when comparing mice to human brain tissue.
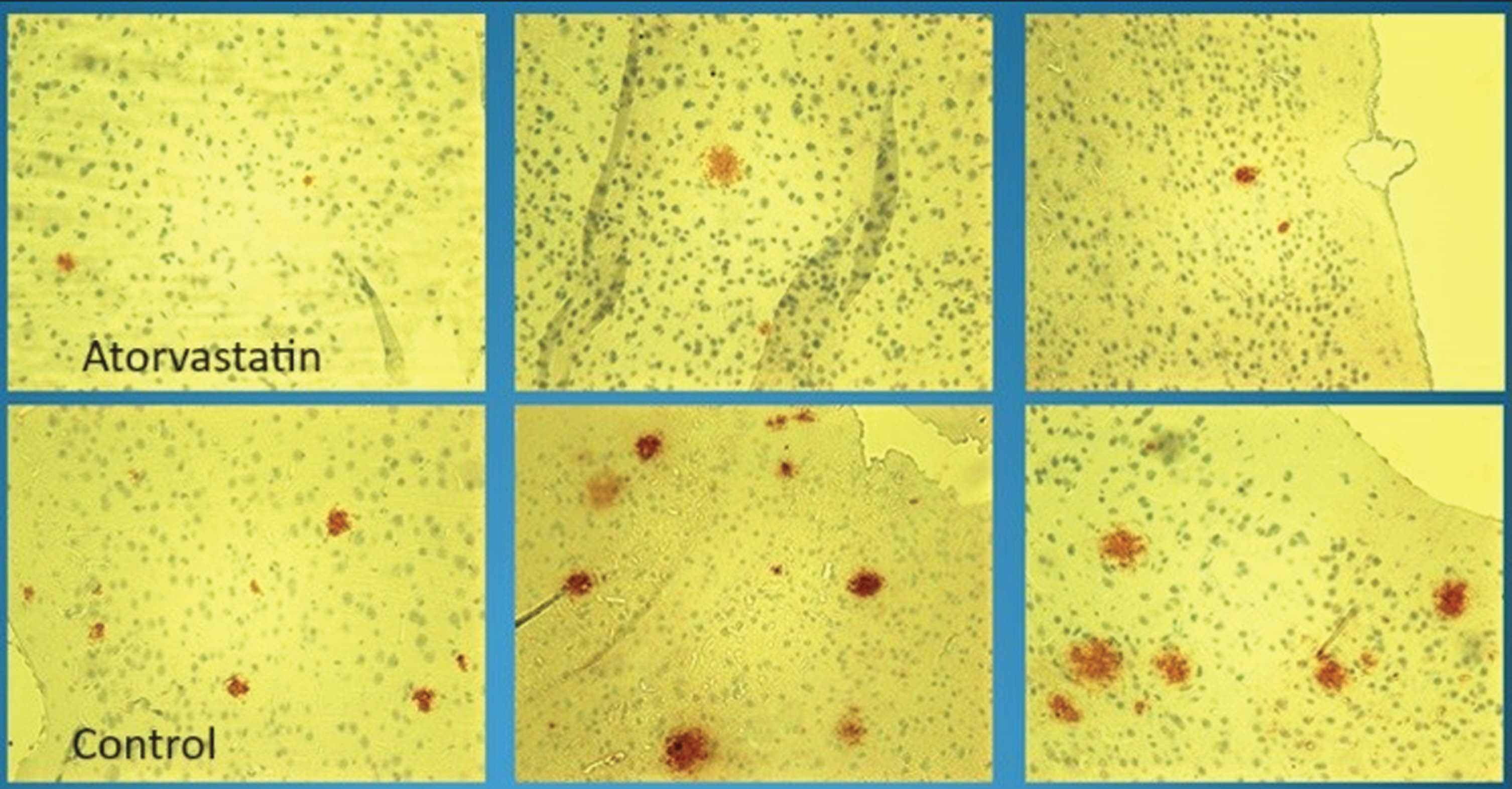
Visualization of Amyloid Plaques in Transgenic Mice via Immunohistochemistry. The depiction compares statin-treated (upper panels) and control mice (lower panels). The statin-treated transgenic mice demonstrated a consistently reduced amyloid plaque burden compared to the controls. Quantitative immunohistochemistry analysis in these experiments was always performed independently and blindly (without knowledge of the treatment groups) by the neuropathologist, ensuring objectivity in the evaluation. “Control” refers to mice not subjected to statin treatment.

Positive correlations were found between the levels of Aβ40 (r = 0.832) and Aβ42 (r = 0.817) peptides in transgenic murine brain tissue and circulating serum cholesterol concentrations. Aβ, amyloid-β protein.

Illustration of the nonlinear relationship between amyloid load and total cholesterol (TC), analyzed using a two-step analytical approach that fit the experimental data. Initially, a linear regression model was applied, followed by a nonparametric regression to capture the nonlinear interplay between amyloid deposition and cholesterol levels. This methodological progression elucidated a heuristic equation characterized by a singular peak flanked by two inflection points.
Applying a nonparametric regression model as a heuristic tool, Pappolla and co-investigators [4] unveiled, in the human brain, a non-linear interaction. Intermediate levels of cholesterol correlated with the highest amyloid deposition. In contrast, very high cholesterol levels inversely hindered amyloid deposition. These findings demonstrate the intricate role of cholesterol in amyloidogenesis in the human brain, among other factors leading to AD progression (Fig. 3). y represents the amyloid load. x signifies the total cholesterol (TC) levels. a0, a1, a2, e1, and e2 are the parameters determined through the regression analysis that characterize the relationship between amyloid load and cholesterol levels.
This original equation captures the dynamics of the interaction between cholesterol and amyloid deposition observed experimentally in the human brain. It highlights the complex nature of their association and is hereby designated the “Pappolla-Herbert equation.”
The role of some of the mentioned factors was also pointed out by data published by Vemuri et al. [17] These researchers showed that “vascular health” variables, other than cholesterol levels, also influenced tau deposition, a critical element in the neuropathological cascade leading to cognitive impairment in patients with AD. In this study, both “vascular health” and amyloid emerged as direct contributors to tau deposition, a marker of neurodegeneration, with the impact of amyloid on tau surpassing that of “vascular health.” Notably, hyperlipidemia was the sole significant predictor of tau deposition among the variables examined. However, their analysis did not directly include the specific effects of cholesterol levels.
Another neuropathological study [18], conducted by Launer et al., sought to understand the association between plasma cholesterol levels (total, HDL, and LDL) and the development of neuropathological markers associated with AD, specifically neuritic plaques (NP) and neurofibrillary tangles (NFT). The study examined a population-based autopsy series of 218 Japanese American men, part of the Honolulu-Asia Aging Study. Cholesterol levels were measured late in life (average age at death 84.6 years) for all subjects, and midlife measurements were available for a sub-sample. The analysis, adjusted for various factors, revealed a significant linear association between increasing late-life HDL cholesterol levels and the number of neocortical NPs and hippocampal and neocortical NFTs. Similar trends were observed for midlife HDL-C levels. This study suggested that the constituents of HDL-C may play a role in the formation of AD pathology. Thus, the findings unveiled a complex interplay between age, genetic predisposition, cardiovascular health variables, and markers of neurodegeneration (amyloid and tau deposition).
Although initial retrospective studies suggested a potential benefit of statins, others found no significant cognitive improvements in AD patients (see below). Unfortunately, these preliminary observations, coupled with an incomplete understanding of the age-related dynamics and other mentioned variables, led to a series of clinical trials, which yielded largely disappointing results. These trials demonstrated critical shortcomings linked to treatment duration, age, follow-up periods, and, most importantly, the stage of AD at which treatment was initiated. These topics will be further analyzed later in this paper.
The brain has the highest cholesterol concentration, carrying approximately 25% of all the cholesterol in the body. Brain cholesterol plays a vital role in several physiological processes, including neurotransmission, synaptic development, and membrane stability [14, 19]. A disturbance of brain cholesterol metabolism could enhance the amyloidogenic Aβ pathway [4, 20], impair brain circulation, and implicate other processes, such as several genetic variables linked to lipid metabolism may be important in the pathophysiology of AD [11, 12]. Several consequential factors in AD pathogenesis emerged, including the roles of cholesterol and oxysterols, apolipoproteins and the metabolism of the amyloid-β protein precursor (AβPP).
THE ROLE OF OXYSTEROLS
Disrupted cholesterol homeostasis encompasses various critical elements from peripheral cholesterol and the de novo synthesis of cholesterol in astrocytes and neurons to the interplay of apolipoprotein E (ApoE), LDL receptors (LDLR and LRP1), and ATP-binding cassette (ABC) transporters [21–23].
In the brain, cholesterol undergoes conversion into oxysterols such as 24-S-hydroxycholesterol (24-OHC), catalyzed by the neuron-specific enzyme CYP46A1 [24]. This conversion is vital to cholesterol homeostasis. Additional roles of 24-OHC include modulation of cholesterol synthesis, cholesterol transport facilitation between astrocytes and neurons, ApoE expression, and prevention of SREBP-1a and SREBP-2 transcription factors’ maturation [25]. The latter role is principally accomplished through its action as a natural ligand for liver X receptors (LXRα and LXRβ) and retinoic acid receptor-related orphan receptors (RORs) [26].
Beyond its critical involvement in cholesterol regulation, 24-OHC has an extensive physiological role in the maturation and survival of nerve cells via its inverse agonist activity towards RORα [27]. Moreover, 24-OHC is a positive allosteric modulator of N-methyl-D-aspartate receptors (NMDARs), an activity that is essential for synaptic plasticity, learning, and excitatory neurotransmission [28].
The complex role of 24-OHC, as the predominant oxysterol in the brain, prompts novel lines of inquiry into potential novel therapeutic targets. However, the intricate roles of oxysterols in the AD brain are yet to be elucidated. Certain oxysterols, such as 27-OHC, 7β-hydroxycholesterol, and 7-ketocholesterol, exhibit a marked increase in AD and have been implicated in disease progression, while 24-OHC levels decline due to neuronal loss [24, 29]. Thus, unexplored areas of discovery and potential therapeutic opportunities still exist.
In a study by Dias et al., the investigators proposed that disrupting the brain’s detoxification capacity for oxysterols via sulfation may impact AD pathogenesis [30]. Upon analyzing lipids from postmortem brain tissue and cerebrospinal fluid from early and late-stage AD patients, the investigators reported increased levels of specific oxysterols (26-hydroxycholesterol, 25-hydroxycholesterol, and 7-oxycholesterol) in late-stage AD brain tissue and mitochondria. The exception was 24S-hydroxycholesterol, which showed a decrease. The authors inferred that these alterations could compromise mitochondrial function in the brain, potentially accelerating AD progression.
Wong et al. advanced the hypothesis that oxysterols play a key role in AD by modulating neuroinflammation [31]. Their data revealed that LPS-induced IL-1β release was amplified by 25-OHC and attenuated by CH25 hydrolase deletion. Moreover, they found that microglia expressing apoE4, an established AD risk factor, produced more 25-OHC than those expressing apoE3 following LPS treatment. They proposed that 25-OHC might influence AD progression by acting as an inflammatory mediator secreted by microglia in the brain, enhancing IL-1β-mediated neuroinflammation in an apoE isoform-dependent manner.
The regulation of cholesterol homeostasis and oxysterol production in the brain and their influence on neuroinflammation extends to other potential factors perhaps involved in AD pathogenesis, such as viral infections. For instance, a study by Gc and colleagues proposed that 25-hydroxycholesterol stimulates innate immune responses during viral infections and activates the integrin-focal adhesion kinase (FAK) pathway [32]. In alignment with the hypothesis of Wong et al. [31], the study established that 25-OHC induces the production of proinflammatory mediators, such as tumor necrosis factor-α and interleukin-6, through direct binding to integrins. This is particularly interesting as it suggests that certain oxysterols may have a broader role beyond cholesterol homeostasis and could contribute to neuroinflammatory processes triggered by specific pathogens, perhaps implicated in AD.
Adding to this narrative are the outcomes of the Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER). This two-year intervention study involving older individuals (60– 77 years) with an increased risk of dementia but without substantial cognitive impairment yielded intriguing results [33]. The intervention, which included a combination of diet, exercise, cognitive training, and vascular risk management, led to a notable reduction in 27-OHC levels in the subjects. This reduction was correlated with cognitive improvement, particularly in memory function. Interestingly, this association was only observed in the intervention group and not in the control group. Moreover, a significant reduction in 27-OHC levels was recorded in those participants with initially high levels of 27-OHC.
Baseline data from the FINGER study also revealed associations between higher 27-OHC levels and lower total gray matter volume, hippocampal volume, and cognitive scores. Although these associations were independent of total cholesterol levels, it is worth noting that gender influenced baseline associations but not the longitudinal ones. This raises the prospect that 27-OHC could serve as a marker for AD risk and be a potential tool to monitor the effects of preventive interventions [33].
The emerging insights from these studies show important relationships between cholesterol homeostasis, oxysterol production, neuroinflammation, oxidative stress, and other potential factors such as infections, genetics, and lifestyle. This understanding will aid in delineating the pathological factors and identifying novel therapeutic targets and prevention strategies.
CHOLESTEROL AND APOLIPOPROTEINS
Apolipoproteins, a class of proteins integral to lipid metabolism, are broadly distributed across a diverse array of vertebrates, including both terrestrial and aquatic species. The evolutionary history APOE is traced back to gene duplications of apolipoprotein C1 (APOC1) occurring approximately 400 million years ago, before the divergence of fish and tetrapods [34]. Remarkably, functional analogs of these proteins have been identified in choanoflagellates, indicating that apolipoproteins represent an ancient protein family that emerged prior to the evolutionary advent of modern animal lineages (Fig. 4). This widespread distribution and deep evolutionary root suggest a fundamental role for apolipoproteins in lipid transport and metabolism across the animal kingdom.

This illustration represents the NMR structure of full-length apolipoprotein E3 (apoE3), determined by Chen et al. [35]. The structure was resolved using solution NMR spectroscopy, providing detailed insights into the molecular conformation of apoE3, a protein critical for lipid metabolism. The image displays the helical regions and overall architecture of the protein, highlighting its structural features. Image credit: Research Collaboratory for Structural Bioinformatics Protein Data Bank.
Human apoE is a major determinant in lipid transport, playing a critical role in atherosclerosis and other diseases. Binding to lipid and heparan sulfate proteoglycans induces apoE to adopt active conformations for binding to the low-density lipoprotein receptor (LDLR) family. ApoE also interacts with the Aβ peptide, exhibiting critical isoform-specific effects.
The NMR structure of apoE3 reveals a unique topology of three structural domains. The C-terminal domain presents a large exposed hydrophobic surface likely to initiate interactions with lipids, heparan sulfate proteoglycans, and Aβ peptides. This topology precisely regulates the tertiary structure of apoE to permit only one possible conformational adaptation upon binding, preventing premature binding to apoE receptors during receptor biogenesis. This ensures optimal receptor-binding activity by fully lipidated apoE during lipoprotein transport in circulation and in the brain [35].
The role of APOE in AD has long been established [36]. It is widely recognized that the presence of the E4 isoform of the ApoE lipoprotein is a significant genetic risk factor for sporadic late-onset AD. The intricate role of ApoE in regulating cholesterol metabolism further emphasizes its importance [37]. ApoE is a lipid carrier in the brain and body, crucial in maintaining cholesterol homeostasis. Therefore, understanding how changes in cholesterol metabolism impact ApoE expression is key to deciphering its role in AD pathology.
Humans possess three primary APOE alleles: E2, E3, and E4 [36]. While the APOE3 allele is considered the reference allele found in most of the population, it is the other two variants that have shown significant association with AD. The APOE4 allele increases the risk of AD in a dose- and age-dependent manner, while the APOE2 allele decreases it. APOE2 homozygotes are estimated to have about a 40% lower risk of developing AD, though this number can vary based on factors such as gender and ethnicity [38] and other genetic influences [39].
Conversely, APOE4 homozygotes face an increased risk of atherosclerosis and AD by 8–12 times. APOE4 carriers with AD have an earlier dementia onset, poorer memory performance, and a higher Aβ burden than non-carriers [40]. The effects of APOE4 on tauopathy, another key hallmark of AD, remain uncertain. Beyond structural pathological changes, APOE4 also seems to exacerbate functional abnormalities of synaptic plasticity and neuronal network connectivity [41].
Several investigators have proposed that restoring some critical ApoE functions in E4 carriers and inhibiting the detrimental activities of ApoE4 may favorably impact AD [42]. The implication of ApoE4 in AD development and its possible modulation served as the subject of extensive research, as reviewed elsewhere [40, 43].
Lipoprotein research and its role in AD development and progression has benefited significantly from using various mouse models such as ApoE-deficient mice, and APOE knock-in mice [44]. However, it is crucial to remember certain key differences in lipoprotein biology between mice and humans. These differences can impact our interpretation and application of ApoE-related findings from mouse studies to humans. For example, in mice, most circulating cholesterol associates with HDL, whereas in humans, most of it binds to LDL [45]. Mice also lack the cholesteryl ester transfer protein (CETP) gene, which plays a significant role in the transfer of cholesteryl esters and triglycerides between lipoproteins [46].
One of the most frequently used mouse models to investigate the function of human ApoE in the central nervous system (CNS) is the human ApoE targeted replacement (TR) mice, developed in Nobuyo Maeda’s laboratory [47]. These ApoE4 TR mice have the endogenous ApoE gene replaced with human ApoE4 and exhibit phenotypes such as altered cholesterol trafficking in the brain, blood-brain barrier (BBB) leakiness, and cognitive deficits [48, 49]. A compelling correlation has been observed across different study models— mouse models of AD, in vitro cell culture models and human data— regarding the effects of apoE isoforms. Each context consistently underscores the detrimental influence of apoE4, as this isoform disrupts various pathways involved in the progression of AD, ultimately leading to dementia. The hierarchy of influence among the isoforms consistently ranks apoE4 as the most impactful, followed by apoE3, and finally apoE2.
The research conducted by Petanceska and colleagues introduces the possibility that the deleterious effects of hypercholesterolemia might partially operate by escalating the expression of apoE4 [50]. These investigators sought to elucidate the relationship between cholesterol and apoE expression by modulating cholesterol levels with diet or pharmacological intervention in a transgenic mouse model of AD [50]. They found that chronic increases or decreases in total cholesterol levels in plasma corresponded with changes in brain apoE mRNA levels and apoE protein expression. Also, cholesterol loading of primary glial cells led to an uptick in cellular and secreted apoE. In contrast, long-term treatment of astrocytes and microglia with statins, which lower cholesterol levels, decreased cellular and/or secreted apoE levels. These findings suggest that a disruption in cholesterol metabolism may elevate the risk of AD, partly due to cholesterol’s impact on the expression of apoE in the brain (Fig. 5), which, in turn, leads to increased amyloid accumulation (Fig. 6).
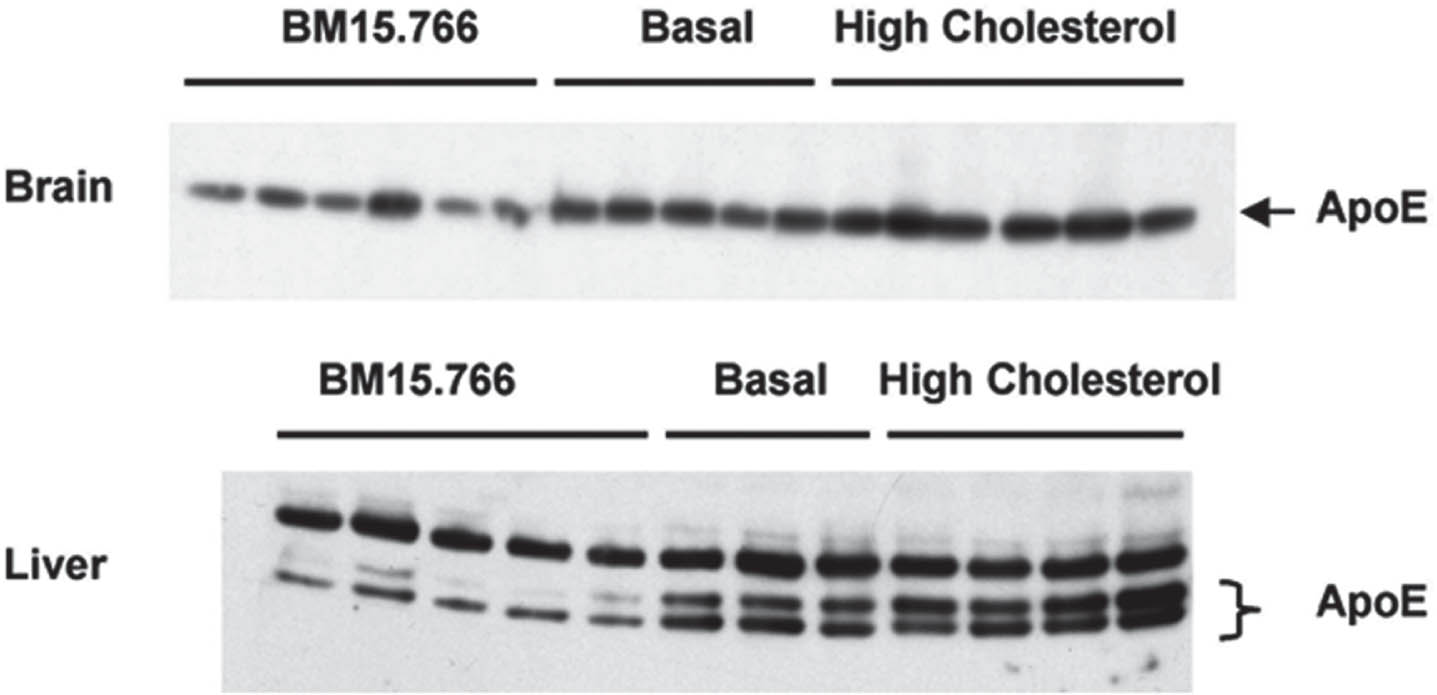
The image illustrates the impact of dietary and pharmacological modulation of cholesterol on apolipoprotein E (apoE) expression in the liver and brain. Brain extracts were prepared using 70% formic acid and then adjusted with 2% SDS/PBS, as previously detailed by Refolo et al. [3] BM15.766 is an inhibitor of cholesterol synthesis. For the western blot analysis, 30 micrograms of protein from both brain and liver extracts were probed using a goat-derived anti-ApoE antibody sourced from Calbiochem in La Jolla, CA. From Petanceska et al. (2003) J Mol Neurosci
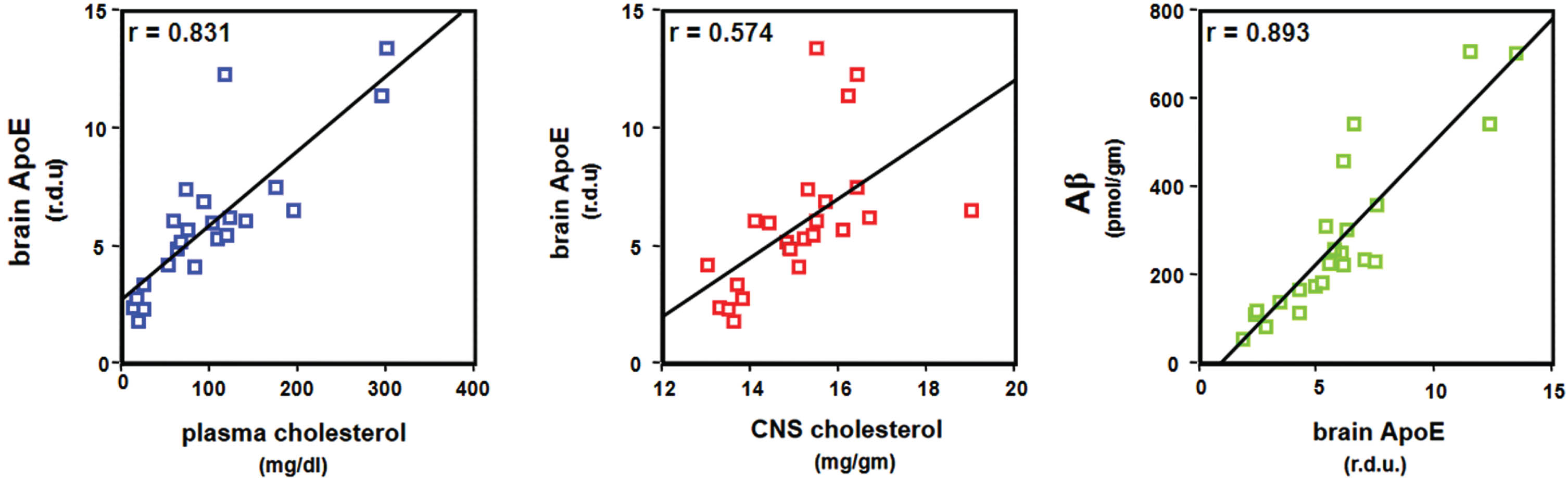
Plasma- and CNS-cholesterol, brain apoE, and brain Aβ in hypercholesterolemic transgenic mice. Graphs illustrate high correlations between plasma and CNS cholesterol, brain levels of apoE, and brain Aβ. From Petanceska et al. (2003) J Mol Neurosci

Molecular structures of statins: simvastatin and pravastatin. This figure illustrates the chemical structures of two commonly used statins: Simvastatin (left) and Pravastatin (right). Simvastatin, a lipophilic statin, has a higher ability to cross cell membranes, including the BBB, and is effective in lowering LDL-c. Pravastatin, a hydrophilic statin, is less likely to cross the BBB but effectively lowers LDL-c levels. The distinct structural differences between these molecules contribute to their varying pharmacokinetic properties and therapeutic effects.
It should be emphasized, however, that the relationship between cholesterol metabolism and ApoE expression is complex and implicates numerous pathways involved in neurodegeneration. Understanding how cholesterol imbalance impacts ApoE functionality and subsequent AD pathology may lead to novel therapeutic targets and a more profound comprehension of disease progression.
CHOLESTEROL AND AβPP PROCESSING
Several theories have been proposed to clarify the apparent correlation between high cholesterol and amyloid accumulation. One theory is that cholesterol might boost the β or γ-secretase enzymes that produce Aβ from AβPP, hinder the α-secretase pathway that is less likely to lead to amyloid formation, or alter other elements such as inflammation or tau metabolism (reviewed in [14]).
Research in animals has suggested that elevated cholesterol could suppress the α-secretase pathway, potentially heightening the risk of AD [2]. This is supported by findings where applying extra cholesterol to human cultured cells overexpressing human AβPP reduced the α-cleavage product of AβPP. Similar results were observed in mice on a high-fat and cholesterol diet [2]. On the other hand, decreasing cholesterol from cultured cells increased AβPP α fragment secretion [51]. Removing cholesterol from hippocampal neurons expressing human AβPP, using treatments like lovastatin and methyl-β-cyclodextrin, also significantly lowered Aβ production, an effect reversible upon reintroducing cholesterol [52].
Additionally, cholesterol levels might influence Aβ aggregation by several mechanisms, including cell membrane alterations [53] or pathological seeding [54].
In conclusion, elevated cholesterol levels may exacerbate AD risk by influencing β- or γ-secretase activity and suppressing the α-secretase pathway, thereby impacting Aβ production and aggregation. Additionally, the conversion of cholesterol into oxysterols, such as 24-S-hydroxycholesterol, can play a substantive role in brain cholesterol homeostasis and has been implicated in AD pathology. The varying levels of oxysterols in AD and their influence on neuroinflammation, oxidative stress and mitochondrial function could exacerbate the risk for developing AD. Understanding these complex interactions is crucial for developing targeted therapeuticapproaches.
CHOLESTEROL AND THE SIGMA RECEPTORS
The sigma (σ) receptors, particularly the σ2 subtype identified as TMEM97, intricately associate with cholesterol metabolism and AD pathophysiology [55]. Hypercholesterolemia may exacerbate AD pathology by modulating the σ2 receptor functions, enhancing their pathological association with Aβ oligomers. It has been shown that the σ2 receptor, in concert with progesterone receptor membrane component 1 (PGRMC1) and low-density lipoprotein receptor (LDLR), forms a complex that regulates the uptake of Aβ oligomers. The interaction between cholesterol, σ2 receptors, and Aβ has been proposed to promote synaptic and neuronal damage characteristic of AD [55].
THE POTENTIAL ROLE OF LDL RECEPTORS
The LDL receptor family, encompassing key members like LDL receptor, LRP1, and VLDLR, was postulated to play a pivotal role in central nervous system health and neurodegeneration, particularly AD [56]. These receptors are integral to synaptic development, endocytosis, and signal transduction within the brain. They modulate cholesterol metabolism in the CNS and play roles in neuronal function and synaptic plasticity. In AD, dysregulated receptor function can influence cholesterol homeostasis and amyloid dynamics, impacting production and clearance. LRP1, for instance, facilitates cholesterol transport to neurons, which is critical for synaptic integrity, while also engaging in the endocytic pathway that influences Aβ accumulation [56].
The evidence suggests that alterations in the function or expression of these LDLR family members could disrupt AβPP processing pathways, thereby augmenting the amyloidogenic processing of AβPP [57]. This LDL-linked mechanism offers another potential therapeutic target, emphasizing the importance of understanding receptor-mediated AβPP trafficking and AD.
In a study by Zambon and collaborators [58], we investigated the incidence of MCI in individuals with familial hypercholesterolemia, a condition characterized by early life exposure to elevated cholesterol levels and LDL receptor dysfunction. Patients with familial hypercholesterolemia showed a significantly higher incidence of amnestic MCI compared to those without familial hypercholesterolemia (21.3% versus 2.9%; p = 0.00). This finding was unrelated to structural brain pathology or white matter disease, suggesting that early exposure to elevated cholesterol or LDL receptor dysfunction is a risk factor. These findings may add evidence on the roles of these receptors in Aβ accumulation. Additional research in this area is essential.
STATINS AND COGNITIVE FUNCTION
Statins are essential in the pharmacological management of hypercholesterolemia and cardiovascular disease prevention. They can broadly be categorized into naturally occurring (Type 1) and synthetic (Type 2) statins. Type 1 statins, such as lovastatin and pravastatin, originated from fungal metabolites and were some of the first members of this class to be utilized clinically. On the other hand, synthetic statins are specifically designed to enhance specific pharmacokinetic and pharmacodynamic properties.
Pharmacologically, statins function by competitively inhibiting HMG-CoA reductase (HMGR), the rate-limiting enzyme in the mevalonate pathway of cholesterol synthesis. This inhibition effectively reduces the synthesis of cholesterol and LDL while modestly increasing HDL levels [59]. The ideal statin exhibits a high affinity for HMGR, selective uptake into hepatic cells, minimal systemic availability, and a prolonged duration of action, reflecting the critical balance between reducing pathogenic lipid levels while maintaining essential cholesterol functions [60, 61].
All statins share a common pharmacophore that mimics the natural substrate of HMGR, but they differ in their ring structures and substituents, affecting their pharmacokinetics and pharmacodynamics [61]. Lipophilicity is a particularly important characteristic that influences a statin’s ability to cross cell membranes, including the BBB, which is pertinent in the context of neurodegenerative diseases like AD [62]. Statin metabolism is primarily hepatic and involves cytochrome P450 isoenzymes, which dictate their plasma half-life, systemic bioavailability, and potential for drug-drug interactions [63].
The bioavailability, potency, and specific affinities for proteins and transport mechanisms vary among statins, contributing to their individual efficacy and side effect profiles. Understanding these properties is necessary for designing statin trials for AD, as they differentially modulate various processes that can lead to neurodegeneration [64]. Choosing a particular statin requires consideration of these characteristics and patient-specific factors such as genetics, comorbidities, tolerance, and overall treatment goals.
In addition, statins may influence cognitive functions through a spectrum of mechanisms, both by directly modulating cholesterol levels and through diverse “pleiotropic” pathways [64, 65]. These agents can disrupt amyloidogenesis and affect tau protein phosphorylation. Additionally, they may enhance endothelial functions and facilitate the removal of neurotoxic factors while diminishing neuroinflammation and oxidative stress [66].
OBSERVATIONAL STUDIES
Many observational studies examined the role of statins in AD prevention (Table 1). In exploring the role of statins in AD prevention or disease modification, it is essential to consider various factors that can influence the study outcomes. These include analytic methods, the age of the participants, the duration of statin use, the specific type of statin employed, sample size, and individual AD risk factors. These elements significantly impact the findings, leading to variable results, from positive to inconclusive or negative. For example, many observational studies (and clinical trials) have been conducted in populations older than 65, overlooking the previously discussed age-related relationship between cholesterol and AD risk [4, 6]. Thus, they missed the “window of opportunity” that would have best captured the potential benefits of these drugs.
Summary of key observational studies on statin use and AD risk or cognitive decline
This table summarizes key observational studies examining the association between statin use and the risk of AD as well as cognitive decline. The key findings highlight both positive associations and null results. The studies are presented in chronological order. AD, Alzheimer’s disease; LLAs, lipid-lowering agents; MCI, mild cognitive impairment; NFT, neurofibrillary tangles; NP, neuritic plaques; MMSE, Mini-Mental State Examination; 3MS, Modified Mini-Mental State Examination.
Despite such diversity, each study contributes unique data. This section reviews representative investigations to understand the implications of the mentioned variables. Due to space limitations, many excellent studies could not be included.
The pioneering investigation on statins and AD was conducted by Ben Wolozin et al. [67], marking one of the first efforts to understand the impact of cholesterol-lowering medications on AD. Utilizing hospital records, the study performed a cross-sectional analysis comparing the prevalence of probable AD among an entire patient population of patients on statins and patients on medications for hypertension or cardiovascular disease. The findings revealed that the prevalence of probable AD was 60% to 73% lower in patients taking statins (specifically lovastatin and pravastatin) compared to the total patient population or those on other treatments. Although this study did not establish causation, it highlighted a potential association between statin use and reduced prevalence of AD, setting the stage for further research. This study was criticized, and the results were partly attributed to reverse causation bias. However, subsequent recent research, as detailed later in this paper, mitigates some of these concerns and highlights alternative explanations.
Having established the conflicting nature of statin research in AD prevention, particularly concerning the age-related dynamics of cholesterol and AD risk, let’s review some individual studies to draw insights from each study’s unique approach and patient demographics.
The Yaffe et al. study, an observational analysis involving 1037 postmenopausal women with coronary heart disease, investigated the relationship between serum lipoprotein levels, statin use, and cognitive function over four years [68]. It assessed how lipoprotein levels and statin treatment changes correlate with cognitive outcomes. Women in the highest quartile for LDL cholesterol exhibited poorer cognitive scores, while those who had reduced LDL levels over the study showed less cognitive impairment. Statin users, including those on simvastatin, atorvastatin, pravastatin, lovastatin, or fluvastatin, demonstrated better cognitive performance than nonusers, suggesting statins’ independent beneficial effect on cognition. The cognitive scores in this study were calculated using the Modified Mini-Mental State Examination (3MS), which evaluates various cognitive functions, including orientation, concentration, language, praxis, and immediate and delayed memory, with scores ranging from 0 to 100. Higher scores indicate better cognitive performance. Participants were classified as having cognitive impairment if their 3MS score was less than 84 points, which is more than 1.5 standard deviations below the mean score of the cohort. Specifically, participants in the highest quartile for LDL cholesterol showed significantly lower cognitive scores (91.9±7.6) than those in the lower quartiles (93.7±6.0), with a p-value of 0.002. They also had a higher likelihood of cognitive impairment, with an adjusted odds ratio of 1.76 (95% CI, 1.04–2.97). Those who showed reduced LDL cholesterol over four years were associated with a decreased risk of impairment, with an adjusted odds ratio of 0.61 (95% CI, 0.36–1.03). Conversely, statin users displayed higher cognitive scores (93.7±6.1 vs 92.7±7.1 for nonusers) and a trend toward reduced cognitive impairment, with an odds ratio of 0.67 (95% CI, 0.42–1.05), suggesting benefits independent of lipid levels.
The study by Zandi et al. [69] examined 4,895 elderly residents (aged 65 years or older) to determine the association of statin use with the prevalence and incidence of dementia and AD. During the three-year follow-up period, out of the initially assessed group, 355 cases of prevalent dementia were identified, with the data indicating an inverse association between statin use and the prevalence of dementia, as reflected in an adjusted odds ratio of 0.44. However, in the follow-up, among 3,308 survivors at risk, 185 cases of incident dementia were identified, and statin use at baseline did not predict the incidence of dementia or AD, nor did statin use at follow-up. The authors concluded that while there might be a lower prevalence of dementia among statin users, there was no clear evidence to suggest that statin use was associated with a reduced subsequent onset (development) of dementia or AD. This research emphasized the challenges of epidemiological studies, including limited follow-up duration, non-specificity regarding statin types, and demography limited to older adults (average age of the participants was 75 years, far beyond the mentioned “window ofopportunity.”)
The study conducted by Rea et al. [70], highlights the complexity of possible outcomes resulting from the type of analyses performed. It encompassed 2,798 individuals aged 65 and older, initially free of dementia. The findings revealed that past statin use did not significantly correlate with a lower risk of various dementia types compared to never using lipid-lowering agents. However, when the authors examined current statin use, the data showed a protective effect against dementia. The investigation revealed that prior statin use, as opposed to current use, did not show a significant correlation with reduced risk of all-cause dementia. These results, demonstrating both positive and negative outcomes, highlight the importance of timing and duration of statin use in addition to other factors.
Jick et al. study [71], encompassed 1,364 participants followed for over six years. It found statins effective in reducing the risk of all forms of dementia. Importantly, the authors examined the effect of statins and other lipid-lowering agents (LLAs) starting at 50 years of age (capturing the “window of opportunity”). Using a nested case-control design, the study utilized data from 368 practices contributing to the UK-based General Practice Research Database. The methodology included three groups of patients who had received LLAs, those with a clinical diagnosis of untreated hyperlipidemia, and a randomly selected group of other individuals. From this base, cases with a computer-recorded clinical diagnosis of dementia were identified and matched with up to four controls on age, sex, practice, and index date of the case. The study included 284 cases of dementia and 1,080 controls. The relative risk estimates of dementia, adjusted for various factors like age, sex, history of coronary-artery disease, hypertension, coronary-bypass surgery, cerebral ischemia, smoking, and body mass index, were near 1.0 and not significant for individuals with untreated hyperlipidemia or treated with non-statin LLAs. However, the adjusted relative risk for those prescribed statins was substantially lower at 0.29 (95% CI 0.13–0.63; p = 0.002), indicating a significantly reduced risk of developing dementia. The interpretation of the study’s results is that individuals aged 50 and older prescribed statins had a significantly lowered risk of developing dementia, regardless of untreated hyperlipidemia or exposure to non-statin LLAs. Despite several limitations, including cross-sectional design and not distinguishing between AD and other forms of dementia, the inclusion of a relatively younger cohort, starting at age 50, might have enhanced the observed beneficial effects of statins, capturing an age group where early intervention could be particularly efficacious in preventing or delaying the progression to dementia.
The study by Li et al. [72], was particularly important because it analyzed the association between statin use and neuropathologic markers of AD, specifically, NP and NFT burden. Despite the small sample size and older age group of the cohort, the study found that statin users had a significantly reduced odds ratio (OR) for each unit increase in the Braak NFT stage compared to non-users (OR 0.44; 95% CI: 0.20 to 0.95). This finding is significant because it shows an association between statin use and reduced NFT and NP, which are important hallmarks of AD pathology. Although there was no significant deviation in odds for each unit increase in the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) staging of NP, the risk for typical AD pathology (Braak stage≥IV and CERAD rating≥moderate) was significantly reduced in statin users (OR 0.20; 95% CI: 0.05 to 0.86). The authors concluded that statins have a protective role against AD-relatedneuropathology.
The study by Masse et al. showed that LLAs may slow cognitive decline in AD patients, suggesting a potential neuroprotective effect [73]. This was an observational study on 342 patients with AD with an average age of 73.5 years and an initial MMSE of 21.3; the study followed them for an average of 34.8 months. Among these patients, 129 had dyslipidemia and were treated with LLAs (47% with statins), 105 had untreated dyslipidemia, and 108 were normolipemic. The study calculated the rate of cognitive decline based on changes in the MMSE score over time and divided patients into slow and fast decliners based on the median annual rate of decline. Results indicated that patients treated with LLAs had a significantly slower decline in MMSE scores (1.5 points/year) compared to patients with untreated dyslipidemia (2.4 points/year) or normolipemic patients (2.6 points/year). Logistic regression analysis further supported the association between LLA treatment and a lower probability of cognitive decline (odds ratio = 0.45, p = 0.002). This study concluded that LLAs, including statins, might confer neuroprotective benefits in slowing cognitive decline among AD patients.
The study by Cramer et al. [74] showed that over a 5-year period, 1,674 older Mexican Americans patients with dementia compared to cognitively normal older Mexican Americans were monitored to assess the relationship between statin use and the onset of dementia and cognitive impairment without dementia (CIND) [74]. Cognitive and clinical evaluations were performed every 12 to 15 months. Statin use was verified through home medicine cabinet inspections. Cox proportional hazards models, adjusted for education, smoking, APOEɛ4 allele presence, and history of stroke or diabetes, were utilized. The study found that 27% (452) of participants took statins during the study period, and of those, statin users were about half as likely to develop dementia/CIND compared to non-users (HR = 0.52; 95% CI 0.34, 0.80). This study did not separate the effect of individual statins.
The study by Haag et al. showed that among 6,992 participants in the prospective, population-based (the Rotterdam Study) followed from 1990–1993 until January 2005, statin use was associated with a decreased risk of developing AD [75]. The study differentiated statin use into any, never used, lipophilic, and hydrophilic categories, with data from pharmacy records and Cox regression analysis adjusting for sex, age, and potential confounders. Over an average follow-up of 9 years, 582 persons developed AD. Compared with never use of cholesterol-lowering drugs, statin use significantly decreased the risk of AD (HR 0.57; 95% CI 0.37 to 0.90), but no significant difference was found with non-statin cholesterol-lowering drug use (HR 1.05; 95% CI 0.45 to 2.44). Both lipophilic and hydrophilic statins showed a decrease in risk, with similar hazard ratios. This study did not separate the effect of individual statins.
Studies with larger samples that allowed examination of individual statins yielded more robust data in favor of statins, particularly simvastatin. In a study by Wolozin et al., the potential benefits of different statins in reducing the incidence of dementia and Parkinson’s disease were explored using data from the US Veterans Affairs database, which includes information on 4.5 million subjects [76]. The study specifically compared the effects of lovastatin, simvastatin, and atorvastatin by employing Cox proportional hazard models to assess subjects on these statins against those taking cardiovascular medications other than statins, adjusting for various covariates related to dementia or Parkinson’s disease.
The study’s key finding was that simvastatin was significantly associated with a reduction in the incidence of dementia in subjects aged 65 years and older across three different models, each incorporating various adjustments such as age, known risk factors for dementia (hypertension, cardiovascular disease, diabetes), and the Charlson index, a measure of chronic disease. Over 700,000 subjects taking simvastatin and over 50,000 subjects taking atorvastatin (aged > 64 years) were analyzed. The hazard ratio for incident dementia was notably lower for simvastatin (HR 0.46, p < 0.0001) than for atorvastatin (HR 0.91, p = 0.11), while lovastatin showed no association with reduced dementia incidence. Additionally, simvastatin exhibited a reduced hazard ratio for newly diagnosed Parkinson’s disease.
The study concluded that simvastatin is strongly associated with a reduction in the incidence of dementia and Parkinson’s disease. In contrast, atorvastatin shows only a modest, non-significant trend in reducing these conditions. These findings highlight the differential impacts of various statins and suggest that specific statins, particularly simvastatin, may offer more substantial neuroprotective benefits.
In a 24-month longitudinal study, Kemp (2020) and colleagues examined the associations between statin use and cognitive changes in older adults [77]. Their study included participants with varying cognitive status, from cognitively normal to AD. Results revealed no significant association between statin use and detrimental cognitive changes or an effect on diagnostic conversion. However, statin use was linked to slower memory decline among those with early MCI. These contributed to the growing consensus that the statins’ potential benefits should be explored in the early stages of cognitiveimpairment.
In a recent 2023 study, Patek et al. explored the impact of statins, on cognitive decline in AD and mixed dementia patients with indications for lipid-lowering treatment [78]. Utilizing data from the Swedish Registry for Cognitive/Dementia Disorders and other national registries, the study compared cognitive trajectories using the MMSE among statin users, non-users, and users of various statin types and non-statin lipid-lowering medications. A particularly significant finding of this study was the observation of a dose-dependent cognitive benefit in patients with AD or mixed AD dementia who were taking statins. Based on longitudinal data from Swedish registries, this study demonstrated that statin users, particularly those using simvastatin, experienced a slower decline in cognitive function as measured by MMSE compared to non-users of statins. Younger simvastatin users showed a more pronounced benefit than younger atorvastatin or rosuvastatin users. The study did not find a significant difference in cognitive decline based on the lipophilicity of the statins. However, the analysis of incident statin users (those who began statin therapy during the study period) yielded inconsistent results, which the researchers suggest could be due to the time-dependent or non-linear effects of statins on cognitive processes or differences in the selection of these users.
Negative observational studies have generally examined older populations. The Arvanitakis et al. study examined the relationship between statin use and AD, focusing on an older population with an average baseline age of 74.9 years [79]. The participants, predominantly women and free of dementia at the start, were part of the Religious Orders Study. Despite the extensive longitudinal data, the study found no significant association between statin use and AD incidence, cognitive change, or AD neuropathology. The older age of the participants might have influenced these findings, considering the potential late-stage intervention of statin therapy, which might be less effective in altering the course of AD or its neuropathological markers. As mentioned throughout this paper, preventative or therapeutic interventions might have a more pronounced impact if initiated earlier in life, addressing risk factors during mid-life. In addition, the study may have underrepresented patients taking brain-penetrant statins, such as simvastatin, which further reduces the ability to detect the effects of statins on AD pathology.
One major criticism against the purported benefits of statins is that the positive results seen in observational studies might be attributed to reverse causation bias. This argument stems from observations that, following a dementia diagnosis, there is a decrease in statin usage among patients. However, a dose-response relationship observed in the Patek et al. study just discussed [78], where increased statin use correlates with more pronounced cognitive benefits, challenges this reverse causation hypothesis. If reverse causation were the primary factor, one would expect to see a uniform decline in cognitive function regardless of statin dosage or duration of use. Instead, the dose-response trend suggests that the protective effects of statins are directly linked to their usage rather than reflect statin discontinuation because of dementia diagnosis.
SYSTEMATIC REVIEWS AND META-ANALYSES OF OBSERVATIONAL STUDIES: THE FDA BLACK BOX
The FDA issued a black box warning on the use of statins in 2012 due to reports of cognitive impairment associated with statin use. These reports included symptoms such as memory loss, confusion, and other cognitive issues. These events were uncommon, and the risk was unclear at that time. However, the Black Box warning initially discouraged patients and practitioners from using statins. Partly prompted by this FDA action, multiple investigators conducted several meta-analyses and systematic reviews. The results of these studies produced cumulative data strongly favoring the use of statins.
Adhikari et al. (2021) conducted a systematic review of studies investigating the association between statin use and cognitive impairment in individuals aged 60 and older [80]. The authors analyzed 24 studies, which included a total of 1,404,459 participants. Of these studies, 21 were prospective observational studies, while three were randomized controlled trials (RCTs). The three RCTs, which had follow-up periods ranging from 3.2 to 5.6 years, showed no significant association between statin use and adverse cognitive effects. The observational studies had follow-up periods ranging from three to fifteen years. Ten of these studies found a reduced incidence of dementia associated with statin use, while seven found no association with incident dementia. Three studies found that cognitive decline was similar regardless of statin use, while one found slower cognitive decline in statin users. The review challenged the FDA black box warning and found no evidence that statin use is associated with adverse cognitive effects, including dementia or decline in global cognition or specific cognitive domains.
In another meta-analysis, Elena Olmastroni (2022) and her colleagues, also motivated by the FDA’s adverse stance on statins, sought to clarify the debated impact of these drugs on cognitive decline [81]. They reviewed observational studies that assessed the risk of AD and dementia in statin users compared to non-users. The researchers searched PubMed, Cochrane, and EMBASE databases up to January 2021 and included cohort or case-control studies reporting AD and/or dementia risk. The results showed that statin use was associated with a reduced risk of dementia (36 studies; odds ratio [OR] 0.80; 95% confidence interval [CI] 0.75– 0.86) and a reduced risk of AD (21 studies; OR 0.68; CI 0.56– 0.81). In a stratified analysis by sex, both men and women showed a similar risk reduction of dementia (OR 0.86; CI 0.81– 0.92). Furthermore, lipophilic and hydrophilic statins were both associated with similar risk reductions. Interestingly, high-potency statins were linked to a 20% reduction in dementia risk, whereas low-potency statins were associated with a 16% risk reduction, although the difference between the two was of borderline statistical significance (p = 0.05). Overall, the study of Olmastroni et al. indicates that statins may have a favorable effect on cognitive health.
Geifman and colleagues (2017) analyzed the potential protective and therapeutic effects of statins in AD from integrated clinical trials and prospective observational studies [82]. The researchers reexamined data from failed AD clinical trials of older individuals. They observed a trend suggesting that simvastatin could slow the progression of cognitive decline, with even more pronounced effects in patients homozygous for APOE4. The study found better cognitive performance among long-term statin users from multiple studies. These observations were further supported by data from an observational cohort, where the incidence of AD was significantly lower among statin users.
A meta-analysis by Poly et al. scrutinized the potential of statins to reduce the risk of dementia by reviewing 30 observational studies from January 2000 to March 2018, with a collective sample of 9,162,509 participants, of whom 84,101 were diagnosed with dementia [83]. The authors found that statin users experienced a 17% lower risk of developing any form of dementia compared to non-users. Specifically, the risk of developing AD was 31% lower among statin users. In contrast, the effect of statins on the risk of developing vascular dementia was not significant. These results offer a compelling counterargument to the notion that reverse causation bias in the context of statin use accounts for the observed risk reduction of AD. As previously mentioned, had reverse causation bias been a significant factor, one would anticipate a uniform effect of statin therapy across all forms of dementia. Instead, the degree of risk reduction observed for AD, as opposed to vascular dementia, indicates that statins exert specific biological effects on the neuropathology of AD.
These meta-analyses revealed no significant association between statin use and detrimental cognitive changes or effect on diagnostic conversion. The findings challenged the FDA’s black box warning on statins causing cognitive deficits and contributed to the growing consensus that statins’ potential benefits should be explored in the early stages of cognitive impairment (Table 2).
Summaries of key meta-analyses examining the association between statin use, dementia or AD risk, and cognitive function
This table summarizes key meta-analyses examining the association between statin use, dementia or AD risk, and cognitive outcomes. The “Key Findings” column summarizes the main results related to statin use and cognitive outcomes or dementia risk. AD, Alzheimer’s disease; OR, odds ratio.
Summary of clinical trials on statin use and AD risk and cognitive decline
This table summarizes key clinical trials examining the association between statin use and the risk of AD or rate of cognitive decline.
CLINICAL TRIALS OF STATINS IN AD
As mentioned previously, epidemiological, preclinical, and observational studies have unveiled three main insights often overlooked in the design of clinical trials for statins in AD. First, elevated cholesterol levels during mid-life is strongly correlated with an increased risk of AD in later years, highlighting the need for early cholesterol management as a potential preventive strategy. Most statin trials (see below) have been conducted in populations older than 65, overlooking the previously discussed age-related dynamics between cholesterol and AD pathogenesis. The potential advantages of statins appear critically linked to the timing of administration, i.e., early intervention— either during the phase of MCI. Secondly, the data favor statin administration either as a preventative strategy in individuals at high risk (hypercholesterolemic individuals or APOE4 carriers). Thirdly, evidence suggests that specific statins, mainly simvastatin, may possess enhanced therapeutic efficacy. At the time of this writing, no trials have addressed these three critical conditionsconcurrently.
While observational studies offer invaluable insights, they come with limitations, such as potential confounding factors and the challenge of causation versus correlation. Addressing these through well-designed clinical trials is crucial, focusing on intervention timing, participant selection, statin types, and genetic and lifestyle considerations. This approach could unveil how personalized cholesterol management— particularly early-life hypercholesterolemia intervention using the appropriate statin— might effectively mitigate AD risk.
For example, the Heart Protection Study (HPS) is often interpreted as showing no statin benefits for cognition [84]. The HPS, a large randomized controlled trial, evaluated the efficacy of simvastatin in reducing cardiovascular events among individuals at high risk for heart disease. Over five years, participants received either simvastatin or a placebo. While the study provided substantial evidence of simvastatin’s effectiveness in reducing heart disease risk, its findings did not conclusively demonstrate benefits regarding the prevention of cognitive decline or AD.
This trial enrolled 20,536 adults from the UK, aged between 40 and 80 years, who had existing coronary disease, other forms of occlusive arterial disease, or diabetes. The participants were randomly assigned to a daily dose of 40 mg of simvastatin over a planned five-year therapeutic timeframe. The study aimed to assess the impact on overall mortality rates and the incidence of fatal or non-fatal vascular events in specific subgroups, alongside secondary evaluations concerning cancer incidence and other significant health outcomes.
The results demonstrated a reduction in all-cause mortality, with 12.9% (1,328 individuals) in the simvastatin group experiencing mortality as opposed to 14.7% (1,507 individuals) in the placebo group, a statistically significant difference (p = 0.0003). This outcome was primarily driven by a notable 18% proportional decrease in coronary mortality rates (p = 0.0005), a slight, yet statistically borderline, reduction in other vascular-related deaths (p = 0.07) and an insignificant decrease in non-vascular deaths. In the HPS, the modified Telephone Interview for Cognitive Status (TICS-m) was employed as a cognitive assessment tool during the final follow-up of participants. This evaluation was conducted either in person at the clinic or via telephone. A TICS-m score of less than 22 out of 39 was predetermined to suggest potential cognitive impairment. As anticipated, lower scores were notably more frequent among older participants and those with a history of stroke.
However, the analysis revealed no significant differences in the prevalence of cognitive impairment between the groups receiving simvastatin and those given a placebo. The proportion of participants deemed cognitively impaired was similar in both groups, 23.7% in the simvastatin group versus 24.2% in the placebo group. This pattern remained consistent across various subgroups, whether differentiated by age at the beginning of the study or by a history of cerebrovascular disease. Additionally, there was no meaningful difference in the average TICS-m scores between the two groups, nor in the incidence of dementia, other psychiatric conditions, or suicide attempts during the follow-up period.
However, the interpretation of HPS as a negative intervention for AD is constrained by several factors, including the specificity of the cognitive measures employed. The HPS utilized the modified TICS-m to assess cognitive function. While this is a validated tool, it might not be sensitive enough to detect subtle changes in specific domains relevant to early AD or to capture the long-term impact of cholesterol management on cognitive decline. Moreover, the HPS did not primarily target cognitive endpoints, particularly in younger cohorts before the initiation of statin therapy, which limits the ability to draw definitive conclusions about the preventative potential of statins against AD.
Finally, and most significantly, the study’s duration (5 years) and the timing of cognitive assessments may not capture the long-term effects of statin therapy on AD risk or progression of cognitive decline, considering the extended preclinical phase of AD and the potential decades-long gap between mid-life cholesterol exposure and the subsequent emergence of clinical AD symptoms. Longitudinal studies with follow-up periods extending 10 to 15 years post-mid-life, focusing on statin administration, would be more indicative of the therapy’s capacity to mitigate later-life cognitive decline or AD onset. Therefore, while the HPS provides valuable data on simvastatin’s cardiovascular benefits, its implications for AD prevention remain unclear.
Another important trial was the LEADe study [85], a randomized controlled trial that evaluated the efficacy and safety of atorvastatin in patients with mild to moderate AD. Participants aged 50– 90, with mild to moderate AD and taking donepezil, were administered atorvastatin 80 mg/day or a placebo for 72 weeks. The study aimed to assess changes in cognition and global function but found no significant benefits of atorvastatin treatment over the placebo.
The study’s approach, although methodologically sound, has several limitations. Firstly, the timing of the intervention might not have been optimal, as intervening at the mild and moderate stages of AD could be too late to observe significant cognitive benefits from statin therapy. Secondly, the choice of atorvastatin and its comparison across different statins is relevant. Not all statins have the same neuroprotective potential, with some evidence suggesting that lipophilic statins like simvastatin could be more effective. Lastly, the study included patients with normal cholesterol levels who might have obscured potential benefits, as statins could have varying effects based on the individual’s lipid profile. The authors addressed some of these limitations in the discussion section of their publication.
Another pivotal study, the randomized controlled trial assessing pravastatin’s impact in an elderly cohort aged 70–82 at risk for vascular disease, aimed to elucidate its effects on cardiovascular health and cognitive function [86]. Conducted over 3.2 years, the trial demonstrated that while pravastatin significantly reduced cardiovascular events, it did not confer any cognitive benefits. This outcome highlights, again, several considerations in statin research for AD, particularly the timing of intervention and the choice of statin. The study’s elderly participants, beyond the optimal mid-life period for cholesterol-lowering interventions to potentially prevent AD, may point to the importance of early preventive strategies. Additionally, pravastatin’s hydrophilic nature, which limits its ability to penetrate the BBB, may render it less effective in mitigating neurodegenerative processes than lipophilic alternatives like simvastatin.
The Sano et al. trial was a randomized, double-blind, placebo-controlled study investigating the impact of simvastatin on individuals with mild to moderate AD, including subjects with normal lipid levels [87]. The study aimed to explore whether simvastatin could slow the progression of AD symptoms. Over 18 months, participants received simvastatin or a placebo, with primary outcomes focused on cognitive changes measured by the ADAS-Cog scale. Despite effectively lowering cholesterol levels, the trial found no significant benefit of simvastatin on cognitive function, global change, or other secondary outcomes. Several factors might contribute to the lack of observed benefit: 1) The trial targeted individuals with mild to moderate AD beyond the early stages, where intervention might have altered the disease’s trajectory more effectively; 2) Participants had normal cholesterol levels, suggesting that their AD pathology might not have been primarily driven by cholesterol-related mechanisms, thus limiting the potential impact of statins. This trial’s results are consistent with other larger studies, suggesting that statin therapy, particularly in patients with normal cholesterol levels and beyond the early stages of AD, does not provide any cognitive benefits.
In a Cochrane systematic review conducted by McGinness and colleagues [88], the researchers assessed statins’ clinical efficacy and tolerability in treating dementia. They identified three randomized controlled trials (748 participants) in which all patients were diagnosed with probable or possible AD. The pooled data showed no significant benefit in cognitive measures, as assessed by the ADAS-Cog and the MMSE. The analysis also revealed no significant treatment-related adverse effects and no evidence that statins were detrimental to cognition. One trail (the ADCLT 2005 trial) indicated that patients with high baseline cholesterol, higher baseline MMSE scores, or the presence of the apolipoprotein E4 allele might maintain better cognitive function on statins, a finding warranting further investigation.
From all these data, the overwhelming evidence from clinical trials is that statins do not show meaningful clinical benefits in slowing AD progression, particularly in older adults or those already diagnosed with AD.
One should point out that exploratory studies on prevention suggest that statins administered to cognitively normal middle-aged subjects at high risk of developing AD may perhaps be modestly beneficial. A study by Sparks and colleagues investigated the association between elective statin use and the reduced incidence of AD in participants of the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT). Analyzing participants who self-reported statin use, the study found a significant decrease in AD risk among statin users after adjusting for demographic and genetic factors. This effect was evident when comparing all users of lipid-lowering agents to non-users. The authors concluded that statin therapy may be of benefit in reducing the risk of developing AD. However, statin users are generally more educated and less likely to smoke [68] (factors that contribute to greater ‘brain reserve’ and may independently protect against AD). Thus, in our opinion, the possibility exists that the apparent benefits of statins in the Starks’ study might be confounded by these lifestyle and demographic variables, suggesting that the statin therapy’s role in reducing AD risk might be overestimated.
In a subsequent study, the same investigators examined the effects of statins on cognitive performance in individuals who had participated in the previous study and had transitioned to MCI. This investigation extended previous data from the ADEPT Trial, highlighting a decrease in AD risk among statin users. However, this benefit did not extend to altering the incidence of MCI. The findings revealed that statin users showed an improvement in delayed recall after converting to MCI, in contrast to those who did not use lipid-lowering agents. This improvement in cognitive function among statin users might underlie the previously observed lowered risk of progressing to AD while maintaining the risk of developing MCI constant. The research thus suggests statins might confer a cognitive protective effect, particularly by enhancing memory recall in individuals post-MCI onset, potentially influencing their conversion to AD. The reasons why statin use modified the risk of developing AD but did not modify the risk for MCI remained unclear. However, one should consider the potential selection bias alluded to above. Also, it is possible that the type of statins evaluated by the studies by Sparks et al. could have contributed to the discrepancies (decreased AD risk but no decreased MCI risk), suggesting the importance of choosing the appropriate statin to maximize potential cognitive benefits.
The study by Carlsson et al. [89], when interpreted in conjunction with another study by Rieske et al. [90], may shed light on this aspect, demonstrating that simvastatin, administered to asymptomatic middle-aged adults at risk for AD, improved certain cognitive functions through specific molecular mechanisms beyond cholesterol metabolism. In a 4-month randomized, double-blind, controlled study, Carlsson et al. evaluated the effect of daily simvastatin (40 mg) versus placebo on cognition in 57 asymptomatic middle-aged adults at increased risk for AD. Compared to placebo, simvastatin improved selected measures of verbal fluency (p = 0.024) and working memory (p = 0.015), independent of APOE4 genotype, gender, and vascular risk factors. In connection with these results, the study by Riekse and colleagues [90] offered insights into certain molecular aspects of such effects. Riekse’s study specifically compared the effects of simvastatin with pravastatin (which has limited CNS penetration) in hypercholesterolemic subjects without dementia. Over a 14-week treatment period, simvastatin significantly reduced phospho-tau-181 (p-tau181) levels in the CSF of all subjects, a form of tau considered a pathological hallmark of AD. No similar reduction was observed with pravastatin, nor were there changes in total tau levels, Aβ peptides (as also noted by Carlsson et al.), soluble amyloid precursor protein (sAβPP) alpha or beta, or F2-isoprostanes. These differential effects highlight the potential significance of some brain penetrant statins in impacting critical molecular markers of AD. Therefore, the timing of statin therapy and the type of statin may both be crucial.
CRITICAL INSIGHTS AND FUTURE DIRECTIONS
This paper presents a comprehensive analysis of the molecular and clinical relationships between cholesterol, specifically hypercholesterolemia, and the risk of AD, alongside the potential therapeutic implications of statins. Observational studies highlight a significant association between midlife hypercholesterolemia and elevated AD risk, advocating for cholesterol management in midlife as a preventive strategy against AD. Conversely, the paradoxical association of high cholesterol levels in older population subgroups with reduced AD risk highlights the intricate role of cholesterol in AD, as shown in the Pappolla-Herbert equation.
All things considered, the overwhelming evidence suggests that while statins still hold a modest promise as a risk-reduction tool in select populations, their overall effect is likely limited.
TRANSFORMING UNEXPECTED OUTCOMES INTO OPPORTUNITIES FOR DISCOVERY
While disappointing, the data from statin trials should be the springboard for novel hypotheses. Although hypercholesterolemia-mediated mechanisms are established risk factors for AD, they may instigate or exacerbate processes that elude statin intervention. For instance, it has been shown that hypercholesterolemia could suppress antiviral cytotoxic T-cell responses [91] and impair antimicrobial immune responses [92, 93] including infections by neurotropic viruses [94]. Hypercholesterolemia can induce changes in oxysterol pathways (see previous section on oxysterols) in a manner impervious to statin therapy. Additionally, the interplay between hypercholesterolemia and LDL receptors [58, 96], or sigma receptors, particularly sigma-2 receptors [55], illustrates other dimensions where cholesterol may influence AD pathology, further complicating our traditional therapeutic thinking and highlighting the necessity for innovative approaches that extend beyond statin intervention.
Future research should not focus exclusively on statins’ preventive potential but dissect the multifaceted nature of cholesterol-related neuropathology, aiming to delineate aspects that novel strategies can effectively target.
AUTHOR CONTRIBUTIONS
Miguel Angel Pappolla (Conceptualization; Data curation; Formal analysis; Methodology; Project administration; Writing – original draft; Writing – review & editing); Lorenzo Refolo (Conceptualization; Data curation; Methodology; Writing – review & editing); Daniel Zambon (Writing – original draft; Writing – review & editing); Kumar Sambamurti (Investigation; Writing – review & editing); Karen Duff (Conceptualization; Writing – review & editing; Collaborated in the research discussed in the paper).
Footnotes
ACKNOWLEDGMENTS
We are deeply grateful to the late Dr. Donald Edmonds Herbert, whose profound expertise in physics, mathematics, and statistics was instrumental in analyzing the data discussed in this paper, including a formula that describes the relationship between cholesterol levels and AD. Dr. Herbert, who passed away on March 4, 2016, had a distinguished career marked by significant contributions to medical physics and statistics, including his work with the US National Cancer Institute and his role as an Emeritus Professor at the University of South Alabama. His legacy in scientific inquiry and education continues to inspire us.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.