
Abstract
Alzheimer’s disease (AD) is a major neurodegenerative disorder impacting millions of people with cognitive impairment and affecting activities of daily living. The deposition of neurofibrillary tangles of hyperphosphorylated tau proteins and accumulation of amyloid-β (Aβ) are the main pathological characteristics of AD. However, the actual causal process of AD is not yet identified. Oxidative stress occurs prior to amyloid Aβ plaque formation and tau phosphorylation in AD. The role of master antioxidant, glutathione, and metal ions (e.g., iron) in AD are the frontline area of AD research. Iron overload in specific brain regions in AD is associated with the rate of cognitive decline. We have presented the outcome from various interventional trials involving iron chelators intended to minimize the iron overload in AD. To date, however, no significant positive outcomes have been reported using iron chelators in AD and warrant further research.
INTRODUCTION
Alzheimer’s disease (AD) is an irrevocable progressive neurological disease manifested by gradual deterioration of memory and cognitive functions. There are various AD hypotheses that include the role of amyloid-β (Aβ) plaques formation, 1 phosphorylated tau propagation, 2 and oxidative stress (OS). 3 Recently, it has been reported that OS occurs prior to amyloid plaque formation and tau phosphorylation, 4 and it is now validated from transgenic mice study. 5 Antioxidant glutathione (GSH) and prooxidant (e.g., iron) imbalance causes unregulated generation of reactive oxygen species (ROS) leading to OS. 6
Disruption of iron homeostasis in microglia promote ROS leading to oxidative damage which plays a dominant role in AD and other neurodegenerative diseases.7,8, 7,8 OS also initiates neuroinflammation and further damage neuronal structures and leads to subsequent neurodegeneration. Studies have shown that iron-loaded microglia are more likely to adopt a pro-inflammatory phenotype, releasing cytokines and chemokines that propagate the inflammatory response and neuronal damage.9,10, 9,10 Activated microglia accumulate iron ions through several mechanisms, including increased expression of iron import proteins such as transferrin receptors and divalent metal transporter 1 (DMT1), and decreased expression of iron export proteins like ferroportin. 8
Iron is found to be the most abundant in the brain, and increased levels of iron are associated with Aβ plaque formation and tau hyperphosphorylation in AD. 11 In vivo study has provided the evidence for the association between iron deposition and tau aggregation in AD. 12 Iron can directly bind to tau, stabilizing its aggregation into neurofibrillary tangles. Iron-mediated OS can lead to the activation of the mitogen-activated protein kinase (MAPK) pathway, including glycogen synthase kinase-3β (GSK-3β) and cyclin-dependent kinase 5 (CDK5), which phosphorylate tau at pathological sites. Hyperphosphorylation of tau is responsible for its loss of normal physiological function leading to aggregation to form toxic.13 –15
Ferritin synthesis is upregulated in response to elevated intracellular iron levels for safely storing iron, thereby preventing iron-induced oxidative damage. Additionally, inflammatory cytokines such as IL-1 and TNF-α can induce the expression of ferritin. In AD, the dysregulation of iron metabolism and chronic neuroinflammation contribute to the upregulation of ferritin expression as a protective mechanism against iron toxicity.16,17, 16,17 Elevated ferritin levels in cerebrospinal fluid or blood can serve as biomarkers for predicting the progression from mild cognitive impairment (MCI) to AD. Monitoring ferritin levels in patients with MCI could provide early indications of AD progression.18,19, 18,19
Various metal ions (e.g., Fe, Cu, Mn, and Zn) play an important role in many physiological processes (e.g., activating signaling pathways, metabolic pathways, etc.). Both iron and copper generate ROS (e.g., H2O2 generates OH– and •OH) through Fenton reaction. However, the biochemical environment in the brain, including the differential distribution of these metals and their binding affinities to various proteins, can influence the outcomes of chelation therapy.
A postmortem study quantifying iron from AD hippocampal tissue found increased accumulation of Fe3+ associated with senile plaque and neurofibrillary tangles formation. 20 Increased iron load and toxicity are associated with a regulated cell death process (ferroptosis). Inefficient antioxidant defense systems, characterized by depleted GSH levels, contribute to ferroptosis due to imbalanced increased lipid peroxidation of polyunsaturated fatty acid (PUFA)-containing phospholipids. 21 The significant increment of loosely bound (non-heme and non-ferritin) iron was reported from the AD patients autopsy samples (frontal cortex and cerebellum) compared to the age matched cognitively normal (CN) subjects. 22 Elevated iron deposition in the brain of AD patients is negatively correlated to the cognitive performance. 23 Furthermore, distinct pattern of the iron levels from hippocampus in MCI and AD patients compared to CN is presented in Fig. 1.

Box plots indicating patterns of iron levels in the brain of healthy group as well as in AD and MCI patients; I) Iron level in the left hippocampus of the autopsy brain of CN of different age groups; 24 II) Iron values from in vivo MRI Quantitative Susceptibility Mapping (QSM) in the left hippocampus (LH) in CN participants of different age groups; 25 III) Iron level in the serum of CN participants from different age groups. 25 IV) Significant elevation of iron in the autopsy brains of AD compared to CN subjects in the hippocampus region; 26 V) Significant increase in the iron level in the AD and MCI subjects as compared to CN as evident from QSM study; 27 VI) Significant depletion of serum iron levels observed in MCI and AD patients in comparison to CN. Serum iron levels are reduced in AD compared to MCI.28,29, 28,29 The respective box plots (II, III, V, and VI) are modified from our earlier work. 28 HC: healthy control; MCI: mild cognitive impairment; AD: Alzheimer’s disease.
GSH depletion has also been correlated with decline in cognitive functions. GSH is primarily used for the neutralization of radicals. 30 The neuroprotective GSH is oxidized to dimeric glutathione disulfide by the accumulation of iron (Fig. 2). The conversion of GSH is demonstrated by NMR spectroscopy from the chemical shift changes of cysteine βCH2 in GSH- iron-sulphur-protein complex 31 (Fig. 2).
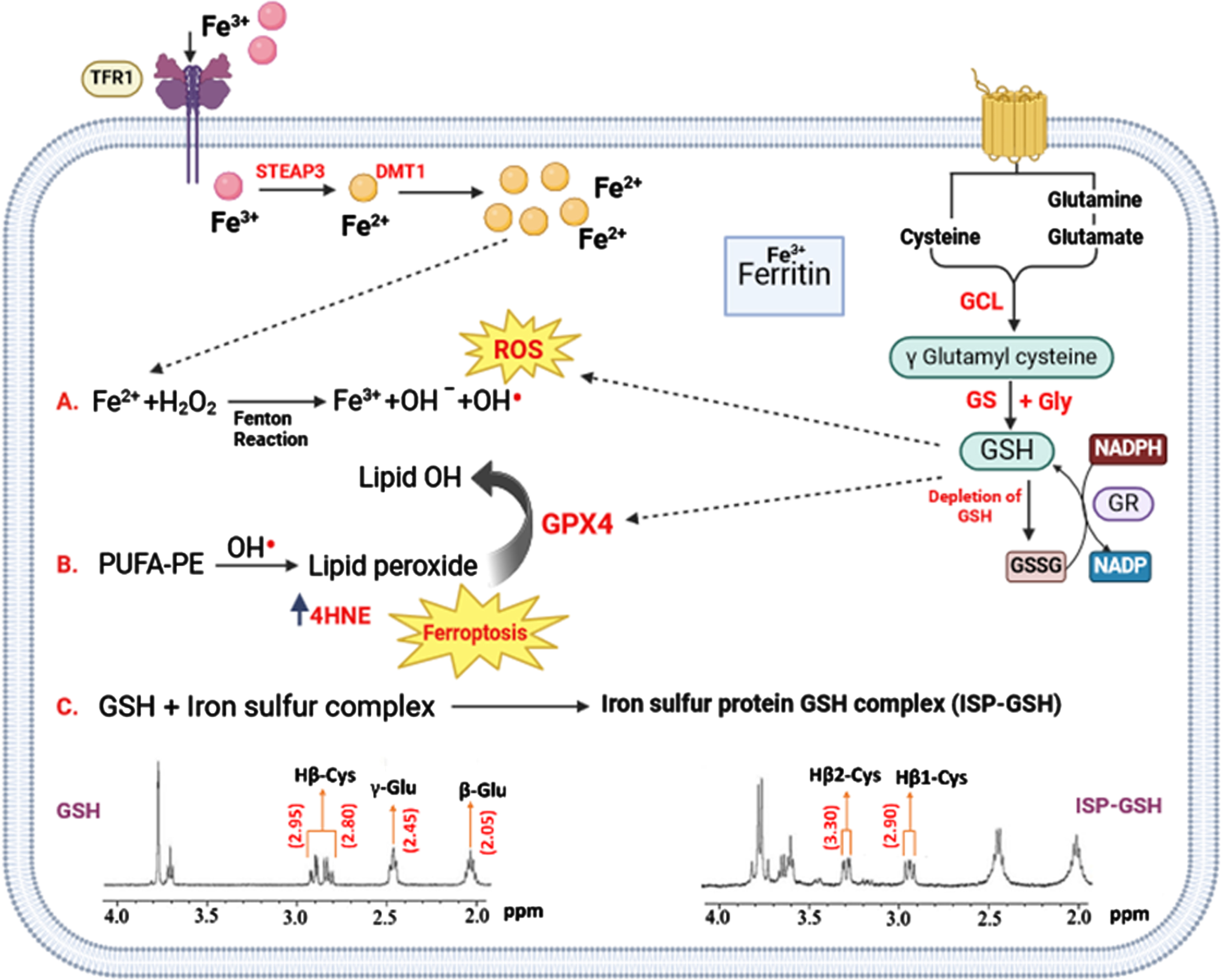
Implication of iron in various processes pertaining to OS in AD. Iron is taken by neurons via TfR1 and subsequently Fe+3 is converted to Fe+2 state by STEAP3. Fe+2 is pumped to cytoplasm by DMT1 and is stored in ferritin in the form of Fe+3 oxidation state. Aging, inflammation, and OS could dysregulate the iron transport proteins and cause iron retention. When GSH decreases in the neurons under severe OS, GPX4 cannot exert the function of anti-lipid peroxidation. PUFA-OOH can be accumulated up to a lethal level to trigger ferroptosis 21 and cell death. Three distinct processes occur in neurons in AD. A) Fenton reaction generates reactive hydroxyl radicals (OH•) and hydroxyl ion through the reaction between hydrogen peroxide (H2O2) and ferrous ions (Fe2+). GSH is being utilized to neutralize ROS. B) PUFA-PE reacts with OH radicals leading to the formation of lipid peroxides, which are converted to lipid-alcohol by the involvement of GPX-4 and GSH is being utilized in this process. C) GSH and iron-sulfur protein (ISP) interact and generate ISP-GSH complex. The NMR spectra of GSH and ISP-GSH complex are presented for clarity, the β-CH2 peak of cysteine amino acid moiety is shifted from 2.80 ppm to 3.30 ppm, indicating the impact of this interaction. Abbreviation: TFR1, Transferrin receptor protein 1; STEAP3, Six-Transmembrane Epithelial Antigen of Prostate; DMT1, Divalent Metal Transporter 1; GCL, Glutamate Cysteine Ligase; GR, Glutathione Reductase; PUFA, Polyunsaturated fatty acids; PE, Phosphatidylethanolamine; ROS, Reactive Oxygen Species; GS, GSH synthetase.
Comprehensive list of clinical trials incorporating DFO, DFP, CQ, and PBT2, along with their respective outcomes. It’s noteworthy that four observational trials reported no significant improvement in AD patients. Quantitative iron mapping of the brain is presented in correlation with these trials, offering valuable insights into the longitudinal variations of iron load in the brain
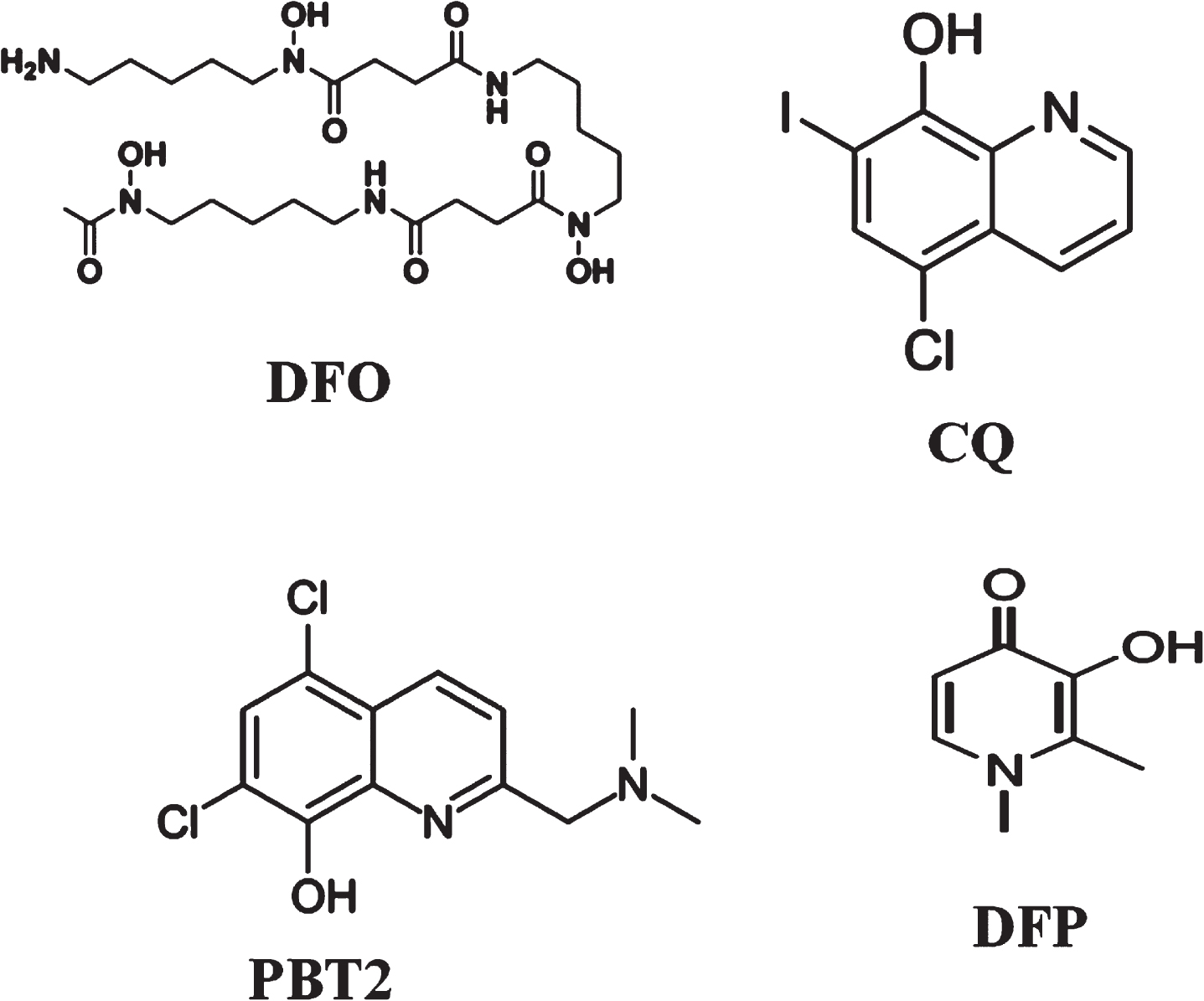
Structures of various iron chelators Desferoxamine (DFO), 5,7-dichloro-2-[(dimethylamino) methyl] quinolin-8-ol (PBT2), Clioquinol (CQ), and deferiprone (DFP). 37 LF is a single-chain polypeptide and carbohydrate moieties with 691 amino acid residues
Both copper and iron can catalyze the formation of ROS through the Fenton reaction, contributing to OS and neuronal damage. However, the biochemical environment in the brain, including the differential distribution of these metals and their binding affinities to various proteins, can influence the outcomes of chelation therapy.
CLINICAL TRIALS IN AD WITH IRON CHELATORS
In AD research, clinical trials involving iron chelators may have potential beneficial effects. There are a limited number of AD trials with iron chelators approved by FDA and these chelators are Desferoxamine (DFO), deferiprone (DFP), 5,7-dichloro-2-[(dimethylamino) methyl] quinolin-8-ol (PBT2), and Clioquinol (CQ) and Lactoferrin (LF). These iron chelators interact with the Fe+3 iron to reduce iron overload in the brain.
DESIRABLE PROPERTIES OF IRON CHELATORS
Since iron overload plays an important role in AD, the use of effective and safe iron chelators to reduce excessive iron is an important avenue for AD patients. A iron chelator should have the following characteristics: 32 1) Iron chelators should be small with high permeability to cross the blood-brain barrier (BBB); 2) Should not have any toxicity ensuring the absence of adverse effects like liver or renal dysfunction, as well as avoiding issues such as bleeding in the brain; 3) It should remove the chelated iron from selected brain areas of interest (e.g., hippocampus, cortical area, etc., in AD) without depleting transferrin-bound iron from plasma; 4) It should transfer iron to other biological proteins, such as circulating transferrin.
TRIAL OVERVIEW
Various interventional studies with iron chelators and observational studies for iron mapping are available from these sources “(https://clinicaltrials.gov/)” and Pan African Clinical Trials Registry.
CONCLUSIONS
The lack of positive outcomes in clinical trials with iron chelators could be attributed to several factors. First, the BBB poses a significant challenge for effective drug delivery to the brain. Many iron chelators have limited ability to cross the BBB, reducing their efficacy in targeting brain iron. Second, the timing of intervention with iron chelators might be critical. Third, patient heterogeneity in terms of genetic background, disease stage, and co-morbidities can influence the response to iron chelation therapy. Finally, the complexity of iron metabolism and its interactions with other metals and proteins in the brain might require a combination approach rather than single-agent therapy.
Autopsy study as well as in vivo QSM studies indicated the importance of iron load in the brain in MCI compared to control subjects. There is only one in vivo study where it has been observed that the transition from CN subject to MCI is accompanied by a significant depletion of GSH levels in the hippocampus and a non-significant increase in iron levels. 27 It is reported that hippocampal GSH level is negatively associated with Mini-Mental State Examination score.
Therefore, future clinical trials should concentrate on brain GSH enrichment in the MCI patients to enhance cognitive performance. Other strategies could be combinational approach involving GSH enrichment agent along with suitable iron chelator.
AUTHOR CONTRIBUTIONS
Pravat K. Mandal (Conceptualization; Data curation; Formal analysis; Investigation; Writing – original draft); Joseph C. Maroon (Methodology; Resources; Validation; Writing – review & editing); Avantika Samkaria (Data curation; Methodology; Validation; Writing – original draft; Writing – review & editing); Yashika Arora (Data curation; Validation; Visualization; Writing – review & editing); Shallu Sharma (Formal analysis; Validation; Visualization; Writing – review & editing); Ashutosh Pandey (Resources; Validation; Writing – review & editing).
Footnotes
ACKNOWLEDGMENTS
Dr. Pravat K. Mandal thanks to Professor George Perry (Editor-in-Chief) to bring this special issue in the long and impactful journey of JAD. Thanks to Ms Beth Kumar (managing Editor) for her support in the review process. Dr. Mandal thanks to National Brain Research Center for providing excellent opportunities to conduct various imaging based research.
FUNDING
Dr. Pravat K. Mandal (PI) thanks for partial financial support from the Ministry of Electronics and Information Technology (Grant 4(5)/2019-ITEA to PKM).
CONFLICT OF INTEREST
Pravat K. Mandal is an Editorial Board Member of this journal but was not involved in the peer-review process of this article nor had access to any information regarding its peer-review.
Authors have no conflict of interest to report.