
Abstract
EphA2 receptor tyrosine kinase fulfils various functions in the development of cancers. Here we analyzed how regulation of EphA2 receptor influences metastatic properties in human melanoma cells in vitro and lung metastasis in vivo. Further, we investigated whether the effects are mediated by Src kinase/focal adhesion kinase (FAK) signaling downstream of EphA2. Therefore, as model Mel-Juso and A375 melanoma cell lines showing different intrinsic EphA2 expression levels were used. To regulate EphA2 expression and activity, we used RNA interference, transgenic EphA2 overexpression, and stimulation of EphA2 activity by adding EphrinA1. Adhesion to fibronectin was increased in EphA2-silenced cells and decreased in EphA2-overexpressing cells. Migration and planar motility were unaffected in Mel-Juso cells, but increased in EphA2-silenced A375 cells and decreased in EphA2-overexpressing A375 cells. Adhesion and migration were unaffected by EphrinA1-stimulation, indicating ligand-independent mechanisms. In vivo we detected increased lung metastasis in mice inoculated with EphA2-overexpressing Mel-Juso cells, substantiating the pro-metastatic effects of EphA2 in melanoma. Activity of Src kinase and FAK were unaffected in EphA2-silenced cells and in response to EphrinA1-stimulation. However, in EphA2-overexpressing A375 cells Src phosphorylation was increased, indicating enhanced Src activity. Together, these data suggest that EphA2 receptor promotes malignancy ligand-independently by mechanisms different from Src kinase/FAK signaling.
Keywords
Abbreviations
erythropoietin-producing hepatocellular carcinoma
Eph family receptor interacting proteins
Eph receptor A2
focal adhesion kinase
sarcoma tyrosine kinase
small interfering RNA
Introduction
Eph receptors represent the largest family of receptor tyrosine kinases. Like their ligands, the Ephrins, they are divided into two groups, class A and class B. In general, EphA receptors bind to EphrinA ligands, which are linked to the membrane via a glycosylphosphatidylinositol (GPI) anchor, and EphB receptors bind to EphrinB ligands, which contain a transmembrane domain and a cytoplasmic tail, although some exceptions exist [1–3]. Due to the capability of the Eph/Ephrin system to signal in a forward or reverse manner and, additionally, to signal by crosstalk with other surface molecules, the Eph/Ephrin signaling is highly complex [4–8] and significantly involved in embryonal development, angiogenesis, but also in cellular transformation, metastasis, and tumor angiogenesis [2, 9–11]. One family member, the EphA2 receptor, was found to be overexpressed in many cancers, such as cancers of prostate, ovaries, bladder, cervix, pancreas, kidney, lung, stomach, breast as well as in glioblastoma multiforme and melanoma [12–23]. Highest levels are mostly found in aggressive cells [15, 25]. EphA2 is known to play a role in cell proliferation and in modification of the cytoskeleton, thereby influencing cell–cell and cell-matrix contacts, and therewith modifying cellular adhesion, migration, and invasion. Of note, in contrast to other receptor tyrosine kinases, EphA2 shows both ligand-independent and ligand-dependent mechanisms [4]. In this regard, EphA2 possesses ligand-independent kinase activity in vitro and, further, EphA2 overexpression significantly enhanced migration of glioma cells in the absence of ligand-stimulation [26, 27]. Moreover, EphA2 is upregulated in Vemurafenib- and Dasatinib-resistant melanoma and uterine cancer cells, respectively, and again, ligand-independent phosphorylation of Serine 897 seems to be a key mechanism [28–31].
The major ligand for EphA2 is EphrinA1 and binding of EphrinA1 induces EphA2 receptor autophosphorylation, followed by internalization and degradation of EphA2 [32]. Miao and colleagues described that activation of EphA2 with its ligand EphrinA1 significantly inhibited the chemotaxis of both vector control and EphA2-overexpressing glioma cells [27]. On the other hand, stimulation with EphrinA1 can promote malignancy as shown by Iida and colleagues, who demonstrated that EphrinA1 promoted the proliferation in a hepatoma cell line [33]. Nevertheless, the effects of EphA2 overexpression or EphA2-stimulation by EphrinA1 on tumor growth and metastasis are not always consistent in in vitro model systems. By contrast, in mouse models a clearer pattern arises. Thus, overexpression of EphA2 [34–36] or induction of EphA2 signaling by EphrinA1 stimulation [37] or EphrinA1 overexpression [38] promoted tumor growth and metastasis. On the other hand, downregulation of EphA2 by siRNA or antisense-cDNA [14, 39] or suppression of EphA2 signaling using EphA2/Fc or small molecule antagonist [40, 41] results in inhibited tumor growth and metastasis. The first description of a signal transduction pathway initiated by EphA2/EphrinA1 was published by Pandey and colleagues who discovered that the p85 subunit of phosphatidylinositol 3-kinase (PI3K) binds to EphA2, leading to increased PI3K activity [42]. Up to now, further signaling molecules and signaling pathways of EphA2 are identified, such as members of the Rho GTPase family and Ras/MAPK pathway [43].
One important mediator of cell adhesion to extracellular matrix is FAK, hypothesized to be a link between Eph receptors and integrins. It was repeatedly demonstrated that stimulation of EphA2 with EphrinA1 leads to dephosphorylation or downregulation of FAK, resulting in inhibition of migration or invasion of prostate, pancreatic adenocarcinoma, or glioma cells [44–46]. On the other hand, EphrinA1 can activate the Src kinase-FAK pathway, leading to RhoA activation and decrease in prostate cell contacts or regulating the re-organization of the actin cytoskeleton, promoting spreading in murine fibroblasts [47, 48]. Although evidence is conflicting as to whether activation of proteins such as FAK, paxillin, p130CAS, and Src-family kinases is up- or down regulated upon ephrin stimulation, in general their involvement correlates with adhesive or de-adhesive cell responses [8]. In our former study we analyzed the effects of X-ray irradiation on metastatic properties and Eph receptor expression in human melanoma cells [49]. We demonstrated that irradiation leads to decrease in EphA2 expression and to anti-metastatic effects, such as decreased motility and migration but increased adhesion. Our results further indicated that EphA2-downstream signaling is mediated, at least in part, by Src kinase and FAK. The aim of the present study was to prove the hypothesis, whether the observed irradiation-induced down-regulation of EphA2 could really account for the observed anti-metastatic effects in melanoma cells with decreased migration and increased adhesion to fibronectin. Accordingly, EphA2 overexpression should result in promotion of metastatic properties in vitro as well as on lung metastasis in vivo. Further, we wanted to analyze, whether the effects are mediated by Src kinase/FAK signaling downstream of EphA2. To regulate EphA2 expression and activity, we used RNA interference (siRNA), transgenic EphA2-overexpressing cells, or induction of EphA2 phosphorylation and hence activation by adding the EphA2 ligand EphrinA1. The experiments were performed in two different human melanoma cell lines, representing a primary tumor (Mel-Juso) or metastatic melanoma (A375).
Materials and Methods
Cell culture and siRNA transfection
The human melanoma cell lines were purchased from ATCC (LGC Standards, A375 cells, CRL-1619), and from the German Collection of Microorganisms and Cell Cultures (DSMZ, Mel-Juso cells, ACC 74). Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum and penicillin/streptomycin (all reagents from Biochrom) at 37°C in a humidified atmosphere with 5% CO2. Cells were tested to be mycoplasma negative using Venor® GeM Mycoplasma Detection Kit (Minerva Biolabs, 11-1100).
For targeted silencing of the EphA2 gene the siGENOME Set of 4 (human EphA2, Thermo Scientific) was used. In preliminary experiments we analyzed EPHA2 gene expression after transfection with the four different EphA2-specific siRNAs. Two siRNAs with the best specific knockdown of EphA2 expression were selected and chosen for further experiments (si-EphA2 #1, target sequence GCAGCAAGGUGCACGAAUU, D-003116-06; si-EphA2 #4, target sequence GCAACGUGAUGUCUGGCGA, D-003116-22). The siGENOME non-targeting siRNA Pool #1 (D-001206-13-05) was used as negative control. 1.5–3.5 × 105 (A375) or 2–5 × 105 (Mel-Juso) cells were seeded into each well of a 6-well plate and allowed to adhere overnight. 0.5 μg siRNA was transfected using DMEM without supplements and 2 μl Lipofectamine 2000 according to the manufacturer’s protocol (life technologies, 11668-027). 24 h after transfection an equal volume of DMEM with 20% fetal calf serum and 2% penicillin/streptomycin was added. EphA2 expression was checked at 24, 48, 72, and 96 h after transfection. For other experiments siRNA transfection was performed in 6 cm- or 10 cm-cell culture dishes. Cell numbers and all reagents were up-scaled accordingly.
Plasmid construction and generation of EphA2-overexpressing cell lines
The eucaryotic expression plasmid containing the human EPHA2 gene (accession no. NM_004431) was purchased from GeneCopoeia™ (vector pReceiver-M60, EX-A0125-M60). The corresponding plasmid for mock transfection (empty vector) was obtained using NotI digestion (New England Biolabs, R0189S), separation of the digested plasmid in agarose gel, and subsequent gel extraction with the illustra™ GFX™ PCR DNA and Gel Band Purification Kit (GE Healthcare, 28-9034-70) according to the manufacturer‘s protocol. Transfection of A375 cells was performed using nucleofector technology as described previously [50]. Mel-Juso cells were transfected in DMEM without supplementals using 1.2 μg plasmid/4 μl Lipofectamine™ 2000 in 24-well plates according to the manufacturer‘s protocol. 24 h after transfection cells were transferred to 6 cm-cell culture dishes in DMEM, 10% FCS, and 1.2 mg/ml G418 was added for selection of transgenic cells. Cell clones were picked, expanded, and GFP-positive cells were sorted using a BD FACSAria II system (BD Biosciences). Stable expression of GFP and EphA2 was regularly tested using FACS (for GFP) or Western blot (for EphA2).
Induction of EphA2 phosphorylation by EphrinA1
3–5 × 106 cells were seeded into 10 cm-cell culture dishes and allowed to adhere overnight. Recombinant mouse EphrinA1 Fc chimera (R&D systems, 602-A1) or purified human IgG (R&D systems, 1-001-A) was added in fresh complete growth medium with a final concentration of 0.3 μg/ml. At the indicated time points, cells were lysed, followed by immunoprecipitation with 800 μg protein/2 μg polyclonal rabbit anti-EphA2 antibody (Santa Cruz, sc-924) as described elsewhere [50]. Subsequently, Western blotting was performed following our former protocol using anti-phosphotyrosine antibody (clone 4G10, Millipore, 05-321) and, as loading control, the anti-EphA2 antibody.
RNA extraction and quantitative real-time RT-PCR
RNA extraction, DNase I digestion, and quantitative real-time RT-PCR was performed as described previously [49]. The primers for EphA2 were 5′–GAAGAGCCCCGTATGCACT-3′(forward) and 5′–GGCTCTCAGATGCCTCAAAC-3′(reverse) and for 18S rRNA 5′–CGGCTACCACATCCAAGGAAG-3′(forward) and 5′–GCTGGAATTACCGCGGCTGCT-3′(reverse). The relative expression of EphA2 was calculated using the delta–delta-CT method versus the 18S rRNA. For each sample two independent experiments each in triplicate were performed.
Protein quantification and Western blot
The preparation of cell lysates, protein quantification with BCA method, SDS PAGE, and Western blot were performed as described previously [49]. For each cell line and treatment at least three independent protein lysates were analyzed. Quantitation of protein bands was performed using TotalLab software (Biostep).
Adhesion to fibronectin
The cells were seeded in 6-well plates and transfected with siRNA as described above. 48 h after transfection the adhesion assay was performed as described previously with each 2×105 cells in 24-well plates [49]. Incubation time for adherence of cells on fibronectin coated wells were 70 min for Mel-Juso cells and 40 min for A375 cells. For samples with siRNA treatment images were taken from single wells prior to lysis of the cells using an inverted microscope (AxioVert 40 CFL, Carl Zeiss). Optical density was calculated by subtraction of the reference absorption from the measured value. From this data the relative adhesion was calculated in proportion to the control (non-coding siRNA, transfection control, or IgG application). Three independent experiments were performed each in triplicate.
Migration assays: Boyden chamber transwell migration assay and scratch assay
To determine the migration of melanoma cells through 8 μm pores a Boyden chamber transwell migration assay was performed. 6×105 cells were plated in 6 cm-cell culture dishes and allowed to adhere overnight. 1 μg siRNA was transfected using DMEM without supplementals and 4 μl Lipofectamine 2000 according to the manufacturer’s protocol. At 24 h after transfection cells were washed with PBS and serum-starved by adding DMEM with 2% bovine serum albumin (BSA). At 48 h after siRNA transfection single cell suspensions were prepared, 2×105 cells were added to the upper compartment of ThinCert™ cell culture inserts (Greiner Bio-one, 662638) and allowed to migrate through the membrane for 24 h under standard culture conditions. Transgenic and untreated cells were also serum-starved 24 h prior to migration assay. EphrinA1-stimulation was performed in untreated cells by adding EphrinA1 Fc chimera or purified human IgG, respectively, in the upper compartment of the chamber. The remaining procedure was performed as described previously [49]. The relative migration was calculated from wells with chemoattractant (10% FCS) proportional to control wells (0% FCS). Three independent experiments each in triplicate were performed.
To determine planar motility of melanoma cells a scratch assay was performed. Cells were seeded in 6-well plates with 8 × 105 (Mel-Juso) or 5 × 105 (A375) cells per well and allowed to adhere overnight. SiRNA transfection was performed as described above. 24 h after transfection an equal volume of DMEM with 20% fetal calf serum and 2% penicillin/streptomycin was added. At 48 h after transfection a vertical scratch was produced in the confluent cell monolayer using a sterile micropipette tip. Wells were washed with PBS and 2 ml/well DMEM with 1% FCS was added. For experiments with EphrinA1-stimulation, EphrinA1 Fc chimera or purified human IgG, respectively, with a final concentration of 0.3 μg/ml was added. For transgenic cell lines and untreated cells for EphrinA1-stimulation, 1.5–2×106 (Mel-Juso), respectively 1×106 (A375) cells were seeded per well. Using an inverted phase contrast microscope, each scratch was photographed at a particular position with 250±15 μm scratch width and the respective x–y-coordinates of the microscope stage were documented. Cells were cultured for another 24 h and photographed again at the same position. The width of each scratch was measured at five defined positions per display window using the AxioVision software package (Carl Zeiss) and the relative scratch width at 24 h after scratch proportional to the first measurement was calculated. At least four independent experiments each in duplicate were performed.
Lung metastasis assay
The animal experiments were performed following a former study [51]. Animal experiments were performed using 9–10 week old female NMRI-nude mice (purchased from the animal breeding facility OncoRay – National Center for Radiation Research in Oncology, Faculty of Medicine and University Hospital, Carl Gustav Carus, Technische Universität Dresden and Helmholtz-Zentrum Dresden – Rossendorf). The animal experiments were carried out according to the guidelines of the German Regulation for Animal Welfare. The protocol was approved by the local Ethical Committee for Animal Experiments (reference number 24-9168.11-4/2012-1). The mice were divided into three groups (each n=10 animals), receiving untreated or mock-transfected Mel-Juso cells or Mel-Juso cells overexpressing EphA2 receptor. The single-cell suspensions were inoculated into the tail vein with 1 × 106 cells/300 μl sterile 0.9% NaCl. A part of the single cell suspensions was used to confirm EphA2 expression using Western blot analysis (data not shown). 13 weeks after injection mice were sacrificed by inhalation of CO2, lungs were dissected, dipped in PBS to remove remaining blood, photographed under standardized conditions using a digital camera (Canon Power Shot G6), and transferred into 4% paraformaldehyde. After 24–48 h, the number of surface metastases was counted under a stereo microscope (Stemi 2000-C, Carl Zeiss).
Statistical analysis
Quantitative data are displayed as mean ± standard deviation (SD). Statistical differences were analyzed by one-way analysis of variance (ANOVA) with post hoc Bonferroni method using OriginPro 8.5G software package (OriginLab). A value of p ≤ 0.05 was defined as statistically significant.
Results
Ectopic regulation of EphA2 receptor
Both used cell lines showed intrinsic EphA2 expression, Mel-Juso at a very low level, A375 cells at moderate level. Transient down-regulation of EphA2 was achieved using siRNA and was shown to be significant at 48 and 72 hours after transfection at mRNA level and up to 96 h after transfection at protein level (Fig. 1A and B). On the other hand, we generated Mel-Juso and A375 cells, stably overexpressing the EphA2 receptor (designated as Mel-JusoEphA2 and A375EphA2, Fig. 1C). EphA2 overexpression remained for at least 10 (A375EphA2) and 15 (Mel-JusoEphA2) cell passages (data not shown in detail). Afterwards, cells were discarded and a new aliquot was used. Activation of the EphA2 receptor was induced by application of the EphrinA1 ligand. The activation was confirmed by immunoprecipitation of the receptor, followed by detection of phospho-tyrosine residues (Fig. 1D). Shortly after EphrinA1 application (30 min) the tyrosine phosphorylation of EphA2 was enhanced, which remained for at least 2 hours. Nevertheless, the enhanced phosphorylation of EphA2 showed transient behavior, at 24 hours after initial EphrinA1 application no EphA2 hyperphosphorylation remained detectable in Mel-Juso cells.

Regulation of EphA2 in Mel-Juso and A375 melanoma cells (A) Relative EphA2 mRNA expression at 24, 48, and 72 h after siRNA transfection with different EphA2-specific siRNAs (si-EphA2 # 1 and si-EphA2 # 4) or non-coding control siRNA (si control). 18SrRNA was used as house-keeping gene. Data are displayed as mean ± SD of triplicates of one out of two independent experiments, *p ≤ 0.05. (B) EphA2 protein expression in Mel-Juso and A375 whole cell lysates was determined by Western blot. Detection of actin served as loading control. Relative EphA2 protein content was quantified by densitometry. Data for EphA2 were normalized to actin and are shown as relative values of the si-control for each time point. Data are displayed as mean±SD (n=3), *p≤0.05. (C) EphA2 protein expression in untreated (MJuntreated, A375untreated) and transgenic cell lines (MJmock, A375mock=transfection control; MJEphA2, A375EphA2=EphA2 overexpression) was determined by Western blot. ECL exposure time for Mel-Juso cells was enlarged to visualize the weak signal also in untreated cells. Detection of actin served as loading control. (D) Phosphorylation of EphA2 was determined by immunoprecipitation and Western blot of Mel-Juso and A375 cells at 30 min, 2 h, and 24 h after IgG- or EphrinA1 treatment and untreated control. Whole cell lysates were immunoprecipitated with anti-EphA2 antibody, followed by Western blot with phospho-tyrosine antibody (clone 4G10), and reprobing with anti-EphA2 antibody to observe total EphA2 content. Representative images from three independent experiments are shown. Relative EphA2 phosphorylation was quantified by densitometry. Data for phospho-tyrosines (p-Tyr) were normalized to EphA2 and are shown as relative values of the untreated control for each time point. Data are displayed as mean ± SD (n=3), *p≤0.05.
In order to investigate the influence of EphA2 regulation on metastatic properties, we analyzed adhesion to fibronectin (Fig. 2A, B), migration through a porous membrane (Fig. 2C), and planar motility (Fig. 2D). Concerning adhesion, the down-regulation of EphA2 (Fig. 2B, left panel) led to a significant increase, by 58% in Mel-Juso and 38% in A375 cells, accompanied by a distinct decrease in EphA2-overexpressing cells, by 24% in Mel-Juso and 58% in A375 cells (Fig. 2B, middle panel). Migration and planar motility was completely unaffected in Mel-Juso cells, after EphA2 silencing, as well as after EphA2 overexpression (Fig. 2C and D, left and middle panel). Nevertheless, in A375 cells we found effects with an increase in migration after EphA2 silencing and decrease after EphA2 overexpression, each around 2.5-fold of siRNA-/transfection control (Fig. 2C, left and middle panel). This is confirmed by analysis of the planar motility in A375 cells, showing increased scratch width, which represents decreased planar motility in EphA2-overexpressing cells (Fig. 2D, middle panel). The planar motility of EphA2-silenced A375 cells was unaffected (Fig. 2D, left panel). Of note, the activation of the EphA2 receptor by adding EphrinA1 showed no effects in both cell lines and EphA2-regulating conditions, neither in adhesion nor in migration and planar motility (Fig. 2B, C, D, right panel).
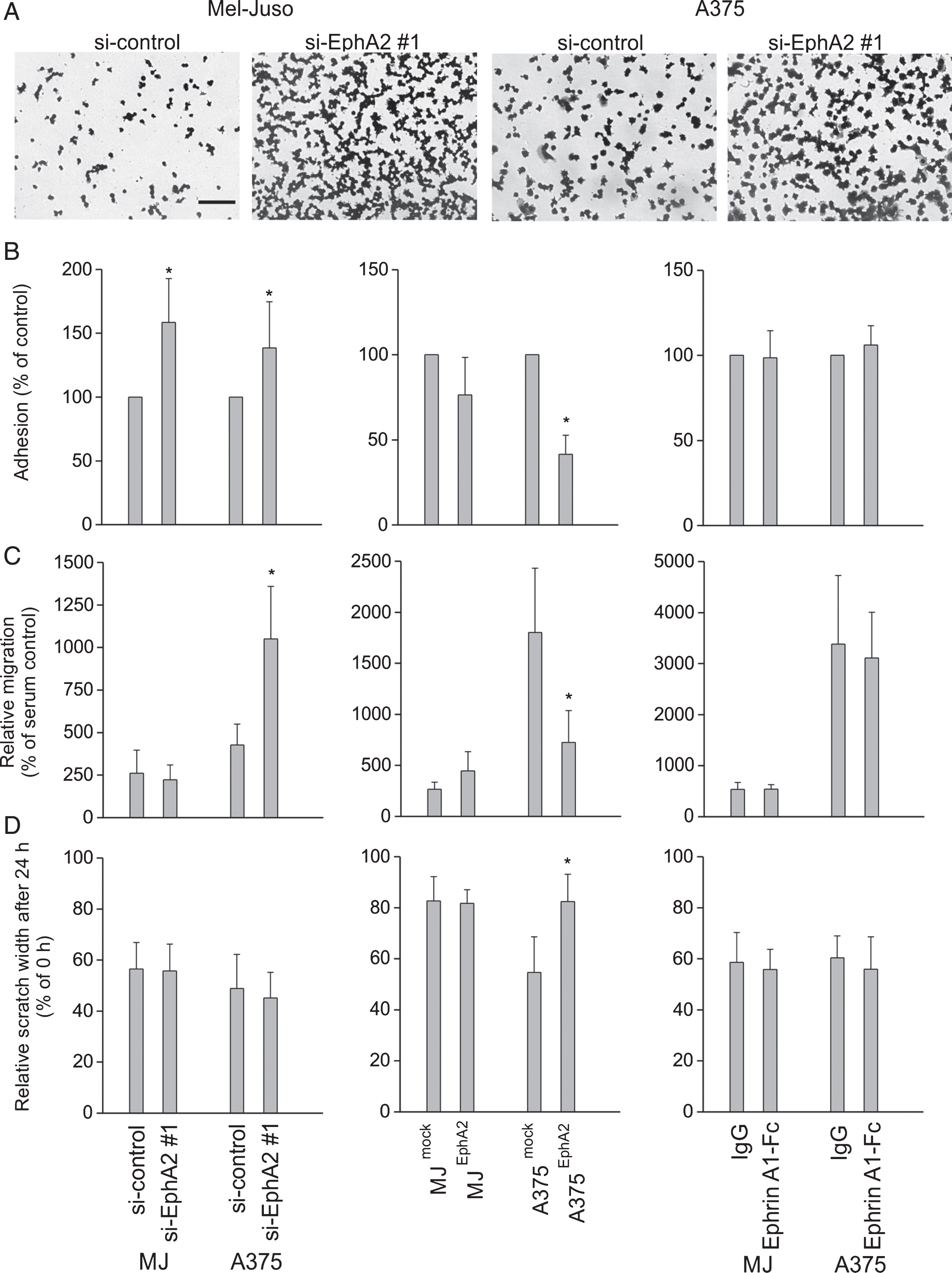
Influence of EphA2 regulation on metastatic properties in Mel-Juso and A375 cells after silencing of EphA2 (left panel B–D), EphA2 overexpression (middle panel, B–D), and EphrinA1 application (right panel, B–D). (A) Representative images of adherent, stained Mel-Juso and A375 cells after application of control siRNA (si-control) or EphA2-silencing (si-EphA2 #1) before lysis and measurement of optical density. Bar=100 μm. (B) Adhesion to fibronectin, relative to the respective control cells. (C) Relative migration of melanoma cells through 8 μm pores determined by a Boyden chamber migration assay. (D) Determination of planar motility using a scratch assay. Data express the relative scratch width at 24 h after scratch generation in proportion to the starting point. (A–C) All quantitative data are displayed as mean ± SD from three or four independent experiments each performed in triplicate, *p≤0.05.
In close accordance to our former study [49], we analyzed the phosphorylation of Src kinase and FAK as important EphA2 downstream mediators. Subsequent to EphA2 silencing as well as after induction of EphA2 phosphorylation, we did not find any relevant differences in the protein content or in phosphorylation of Src or FAK (Fig. 3A and C). Similar result applied after EphA2 overexpression in Mel-Juso cells (Fig. 3B, left panel). However, in EphA2-overexpressing A375 cells we reproducibly detected an increase in phosphorylation and protein content of Src, but no significant changes in FAK protein content or phosphorylation (Fig. 3B, right panel).
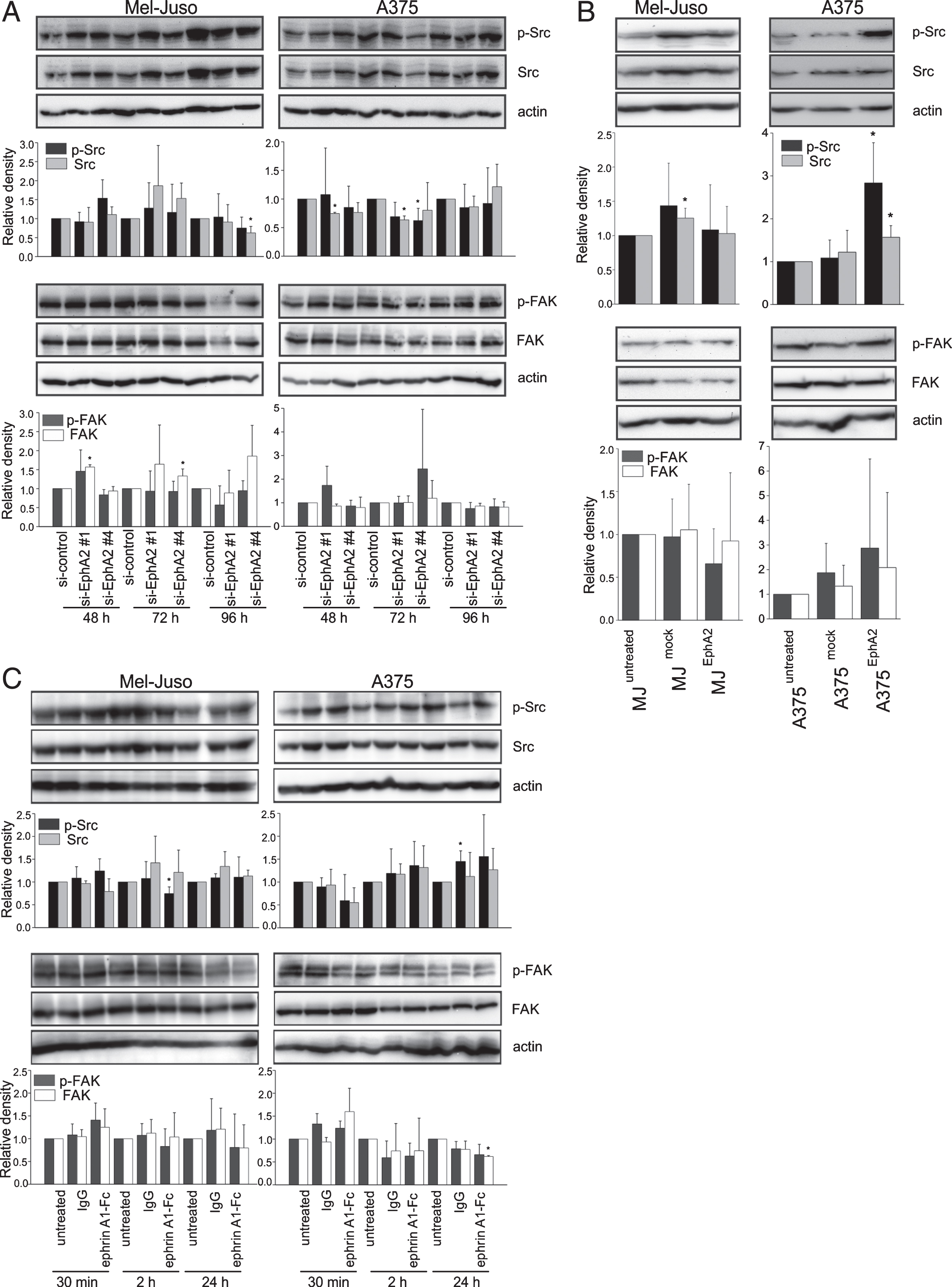
Detection of total Src kinase/FAK protein and phosphorylated, and hence, activated, Src/FAK (p-Src, p-FAK) by Western blot. Detection of actin served as loading control. Quantitation was performed by densitometry. Data for p-Src/p-FAK and total Src/FAK were normalized to actin and are displayed as relative values of the control ((A) si-control, (B) and (C) untreated cells) for each time point. Data are displayed as mean ± SD (n=3), *p≤0.05. (A) Representative Western blot images and quantitation of Mel-Juso and A375 cells at 48 h, 72 h, and 96 h after silencing of EphA2. (B) Analysis of untreated (MJuntreated, A375untreated) and transgenic cell lines (MJmock, A375mock=transfection control; MJEphA2, A375EphA2=EphA2 overexpression). (C) Representative Western blot images and quantitation Mel-Juso and A375 cells at 30 min, 2 h, and 24 h after IgG- or EphrinA1 treatment and untreated control.
Additionally, to analysis of effects of EphA2 regulation on metastatic properties in vitro, we performed a pilot experiment employing a lung metastasis assay in mice. For this experiment we selected EphA2-overexpressing Mel-Juso cells. In preliminary experiments we were able to generate lung metastases in NMRI nude mice at about 12–14 weeks after intravenous (i.v.) injection of Mel-Juso cells, but not after i.v. injection of A375 cells (unpublished results). Further, injection of EphA2-silenced cells was not considered, because down-regulation using siRNA, as performed for the in vitro experiments, remains too short for in vivo experiments with several weeks necessary for sufficient tumor/metastases growth.
In general, metastasis load after i.v. injection of Mel-Juso cells was very heterogeneous ranging from 0 to 43 lung surface metastases. Most mice showed no or only few metastases. However, about one fifth (6 out of 30 animals) showed a high metastasis load. Representative images of dissected lungs are shown in Fig. 4B and C. Comparing the different mouse groups, we found a clear trend: mice, inoculated with untreated Mel-Juso cells, showed no or only up to 5 metastases (designated as low or medium metastasis load). In the second group with mock-transfected Mel-Juso cells, eight animals showed no or few metastases, but two animals had a high metastasis load. In the third group, inoculated with EphA2-overexpressing Mel-Juso cells, only two mice had no metastases (Fig. 4A). The remaining eight mice showed medium or high metastasis load (4 mice each). Taken together, our data support the promoting role of EphA2 in melanoma metastasis.
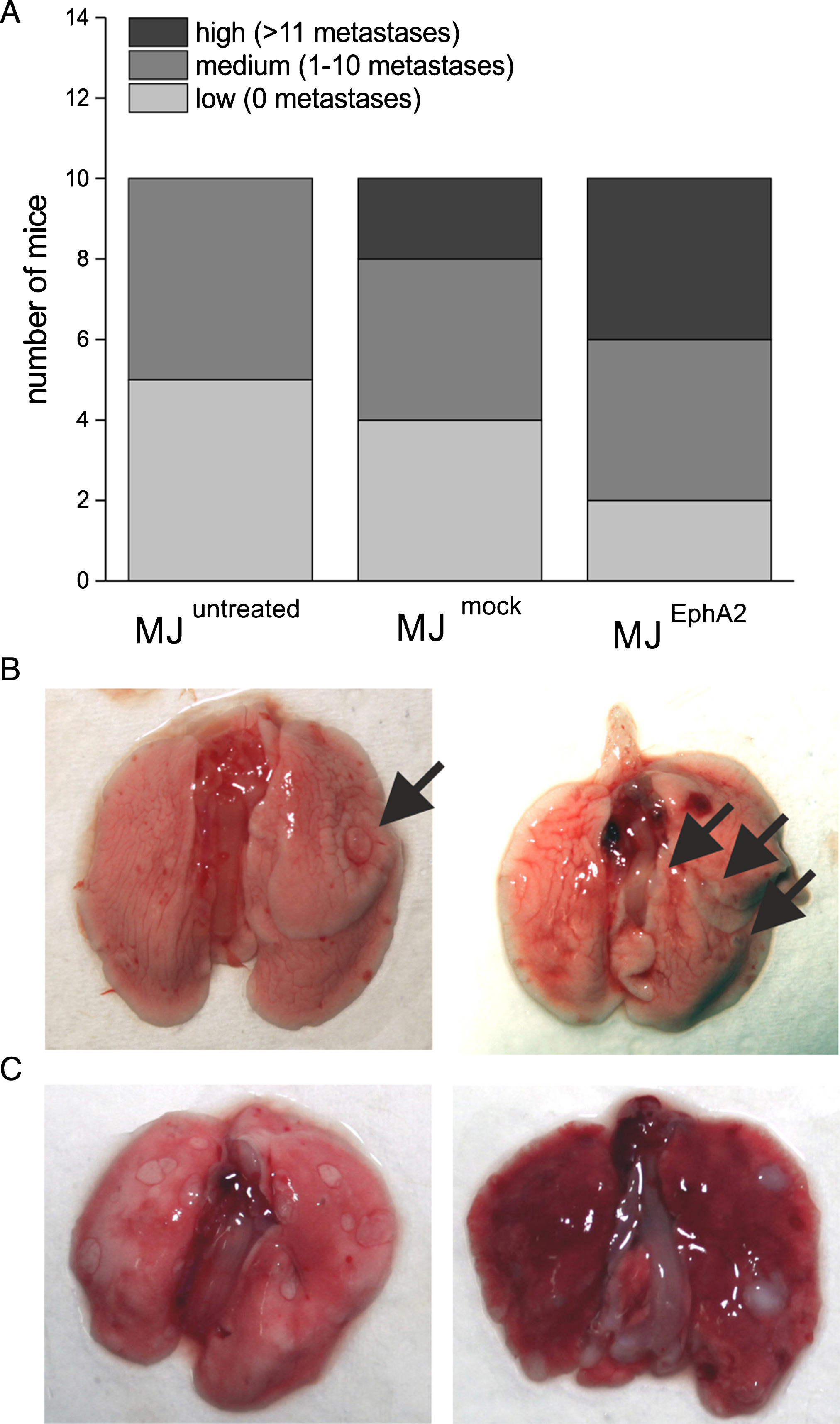
Lung metastasis after i.v. injection of untreated, mock-, or EphA2-transfected Mel-Juso cells. (A) Mice were divided into three categories according to number of their lung metastases: “low” (all data below the 20th percentile, no metastases), “medium (within 20th and 80th percentile, 1–10 metastases), and “high” (all data above the 80th percentile, 11 and more metastases). Representative images of mouse lungs with (B) “medium” and (C) “high” metastasis load.
In our former study we demonstrated that X-ray irradiation of melanoma cells leads to decrease in EphA2 expression and activity (phosphorylation) and to anti-metastatic cellular effects, such as increased adhesion but decreased motility and migration. We supposed that the observed effects, at least partly, were mediated by EphA2 signaling via Src kinase and FAK [49]. Subsequently, we now investigated, if indeed the irradiation-induced EphA2 down-regulation is responsible for the observed cellular effects. In this regard, a reduction of EphA2 should result in increase of adhesion and decrease of migration and vice versa, EphA2 overexpression should lead to decreased adhesion and increased migration.
In this study, the regulation of EphA2 expression was successfully accomplished using siRNA for significant reduction of the EphA2 receptor. Furthermore, two transgenic A375 and Mel-Juso cell lines, stably overexpressing EphA2 successfully were generated. Concerning adhesion to fibronectin the above assumption was substantiated. In our in vitro experiments, we detected a significant increase in adhesion to fibronectin in both Mel-Juso and A375 melanoma cells after silencing of EphA2 and a decrease in EphA2-overexpressing Mel-Juso and A375 cells. Relating this result to our former finding of irradiation-induced down-regulation of EphA2 accompanied by increased adhesion, our recent data support the hypothesis that irradiation affects cellular adhesion via the down-regulation of the EphA2 receptor. Furthermore, it substantiates a pro-metastatic effect of EphA2 overexpression in melanoma as described by other authors [25, 52]. When increasing the phosphorylation and thus activity of EphA2 by application of EphrinA1, we found no significant effects on cellular adhesion. This is clearly indicative for a ligand-independent mechanism, by which EphA2 receptor affects adhesion. This could represent an alternative signaling pathway to the previously reported dependence of inhibition of integrin-mediated cell adhesion from EphA2 kinase activity in prostate carcinoma [53].
As further metastatic properties we analyzed migration by Boyden chamber assay and planar motility using a scratch assay. In both assays, Mel-Juso cells, either EphA2-silenced or EphA2-overexpressing cells, showed no significant changes. By contrast, in A375 cells we found an increase in migration in EphA2-silenced cells and, vice versa, a decrease in migration and motility after EphA2 overexpression. However, despite of the observed heterogeneity in melanoma cell lines used, for A375 cells data concerning migration/motility are consistent between the two regulatory experimental conditions with EphA2 overexpression or EphA2 silencing and would support a scenario with anti-metastatic effects of EphA2 in melanoma. Moreover, in our former study [49] we demonstrated EphrinA1 mRNA expression in A375, but not in Mel-Juso, which would be in accordance with the above mentioned effects when underlying the diametrically opposite roles of EphA2 for cell migration found by Miao [27]. However, these findings are contrary to our former data and to data from the literature. In the former study we detected irradiation-induced down-regulation of EphA2 expression and further, irradiation-induced down-regulation of migration. Thus, we hypothesized, that irradiation-induced down-regulation of EphA2 is responsible for the observed anti-metastatic effects such as decreased motility and migration. By conversion, our former data supported the pro-metastatic role of EphA2 in melanoma as also described by other authors [25, 54]. For instance, Udayakumar and colleagues analyzed EphA2-silenced MM455 and WM115 melanoma cell lines and detected decreased transwell migration [52]. They further identified EphA2 as an essential survival factor in MM455, WM115, and UAC903 melanoma cells. However, cell survival was not analyzed in our study, but of course, could also influence metastatic properties. However, it is also conceivable that, on the basis of changes in EphA2 receptor amount alone, rather adhesion is influenced than migration in melanoma cell models. Furthermore, when analyzing the induction of EphA2 activity by EphrinA1 application, again, as for adhesion experiments, no significant changes were found. This leads to the assumption that the observed effects of EphA2 receptor on migration and planar motility are also caused by ligand-independent mechanisms. Possibly, this depends on tumor entity, because other groups, analyzing prostate, glioma, and pancreatic adenocarcinoma cells, reported an EphrinA1-mediated, and hence ligand-dependent inhibition of migration [27, 53].
However, the presented data were generated using in vitro systems lacking a “natural” environment with other cells and extracellular matrix components, forming the basis for successful dissemination of tumor cells and formation of metastases. Therefore, we analyzed the influence of EphA2 expression in melanoma cells on the formation of lung metastases in vivo. In mice, injected with EphA2-overexpressing Mel-Juso cells, we detected lesser animals with a low or medium metastasis load, and 40% of the animals with high metastasis load. This reveals a substantially higher metastases load than in the control groups with 80–100% animals with low and medium metastasis load and 0–20% high metastasis load. Taken together, our data from the animal model support the hypothesis of a pro-metastatic role of EphA2 in melanoma progression and metastasis.
According to our former study, we also analyzed the expression und activation of Src kinase and FAK, two important down-stream targets of EphA2 receptor. Of note, neither EphA2 silencing nor EphA2 stimulation by EphrinA1, nor EphA2-overexpression in Mel-Juso cells showed any considerable changes in Src/FAK expression or phosphorylation. However, the overexpression of EphA2 in A375 cells reproducibly revealed an increase in the protein content and also phosphorylation of Src kinase, indicating Src activation by high levels of EphA2 receptor. Comparing the present data with our former results, we could not clearly substantiate our hypothesis that anti-metastatic effects in irradiated melanoma cells are mediated by EphA2 signaling via FAK and Src kinase [49]. Merely in A375 cells we detected an increase in Src protein and phosphorylation in EphA2-overexpressing cells, suggesting an activation of EphA2-Src kinase signaling. Of note, this signaling pathway was not proved for Mel-Juso cells, which could indicate that increasing the EphA2 level in cells with (intrinsic) relatively high EphA2 expression (A375) could have different effects than increasing the EphA2 level in cells with (intrinsic) relatively low EphA2 expression (Mel-Juso). The relation between EphA2 receptor and Src kinase has been substantiated by other authors. Knoll and Drescher identified Src family kinases as important components of EphA receptor-mediated axon guidance [55]. Parri et al. provided evidence that EphrinA1 stimulation leads to the recruitment and activation of a Src-FAK complex in PC3 cells [48]. Further, Fang et al. detected significantly higher levels of Src kinase, associated with EphA2 receptor in breast cancer cells overexpressing EphA2 compared to controls, although, Src kinase expression and tyrosine phosphorylation were not significantly changed [56].
The relation of EphA2 receptor and FAK is well described in prostate and pancreatic cancer. Repeatedly, it has been demonstrated, that activation of EphA2 receptor by stimulation with EphrinA1 leads to a dephosphorylation of FAK [44, 57]. On the other hand, silencing of EphA2 by siRNA increased the phosphorylation of FAK in PC3 prostate cancer cells [58]. Nevertheless, in our study we could not clearly affirm this relation. Neither in EphA2-silenced cells nor in EphA2-overexpressing cells or after EphrinA1 stimulation, we detected relevant changes in FAK expression or phosphorylation. However, alternative signaling pathways, such as signaling through small GTPases of the Rho family (Rho, Rac, and Cdc42), Ras family (culminating in phosphorylation and activation of the MAP kinase) or PI3K [4, 5] could mediate the observed effects of EphA2 on melanoma cell migration and metastatic properties.
Taken together, we could demonstrate that regulation of EphA2 receptor influenced cellular adhesion in a pro-metastatic manner, but migration and planar motility in an anti-metastatic manner in vitro in melanoma cells. The observed effects were ligand-independent. In vivo, EphA2 overexpression leads to increased lung metastasis, confirming the pro-metastatic role of EphA2 in melanoma. Regarding our former hypothesis, that the observed irradiation-induced anti-metastatic effects in melanoma cells are mediated by down-regulation of EphA2, we conclude, that the portion of EphA2 signaling is marginal and alternative signaling mechanisms presumably play a major role. Further, the signaling of EphA2 receptor via Src-FAK axis could only be proved for EphA2-overexpressing A375 cells, having a “very high” level of EphA2 expression.
Declaration of interest
No potential conflicts of interest were declared.
Footnotes
Acknowledgments
The excellent technical assistance of Catharina Heinig, Mareike Barth, Aline Morgenegg, and Regina Herrlich is greatly acknowledged. The authors also thank Bettina Reissenweber, PhD, for the generation of the A375EphA2 and A375mock cells. The authors thank the Helmholtz Association for funding a part of this work through the Helmholtz Cross-Programme Initiative “Technology and Medicine – Adaptive Systems”. Christin Neuber and Jens Pietzsch also are thankful to the Deutsche Forschungsgemeinschaft (DFG) for supporting this work within the Collaborative Research Center Transregio 67 “Functional Biomaterials for Controlling Healing Processes in Bone und Skin – From Material Science to Clinical Application“ (CRC/TRR 67/3).