
Abstract
DNA molecules are increasingly being synthesized in the laboratory. A major concern in this domain is that a malicious actor can potentially tweak a benevolent synthesized DNA molecule and create a DNA molecule with harmful properties (Biodefense in the Age of Synthetic Biology (2018) The National Academies Press). To detect if a synthesized DNA molecule has been modified from the original version created in the laboratory, the authors in (In Proceedings of the New Security Paradigms Workshop (2018) ACM) proposed a digital signature protocol for creating a signed DNA molecule. However, several challenges arise in more complex molecules because of various forms of DNA mutations as well as size restrictions of the molecule that impact its properties. The current work extends (In Proceedings of the New Security Paradigms Workshop (2018) ACM) in several directions to address these problems. A second concern with synthesized DNA is that it is an intellectual property. In order to allow its use by third parties, an annotated document of the molecule needs to be distributed. However, since the molecule and document are two different entities, one being a physical product and the other being a digital one, ensuring that both are distributed correctly together without tampering is challenging. This work also addresess this problem by transforming the document into a DNA molecule and embedding it within the original molecule together with the signature.
Keywords
Introduction
Synthesizing DNA molecules in the laboratory is quite common these days. Such a synthetic DNA molecule is often a licensed intellectual property. DNA samples are shared between academic laboratories, ordered from DNA synthesis companies and manipulated for a variety of purposes, for example, to create new biochemicals, reduce the burden of diseases, improve agricultural yields or simply to study the DNA’s properties and improve upon them. There have also been instances of new biological organisms that do not exist in the natural world being created using synthesized DNA [17]. While the vast majority of such activities are pursued for beneficial purposes, there are concerns that malicious users can use the technology malevolently, for example, to make harmful biochemicals, or render existing bacteria more dangerous [17]. Recently, a DNA-based security exploit was demonstrated as a proof-of-concept, where a synthesized DNA was used to attack a DNA sequencer that has been deliberately modified with a vulnerability [18]. Preventing such malicious use of synthesized DNA is beyond the scope of this current work. However, attribution of a physical DNA sample and establishing proof of origin can contribute significantly to deter such malicious activities.
Following the anthrax attack of 2001, there is an increased urgency to employ microbial forensic techniques to trace and track agent inventories. For instance, it has been proposed that unique watermarks be inserted in the genome of infectious agents to increase their traceability [12]. The synthetic genomics community has demonstrated the feasibility of this approach by inserting short watermarks into DNA without introducing significant perturbation to genome function [6,8,15,24]. The use of watermarks has also been proposed in order to identify genetically modified organisms (GMOs) or proprietary strains. Heider et al. [7], for example describe DNA-based watermarks using DNA-Crypt algorithm. This technique is applicable to provide proof of origin to a DNA molecule. However, there is a major shortcoming with all watermark based approaches. The watermark in all these works is generated from an arbitrary binary data and added to the original sequence, and so is independent of the original sequence and provides no integrity of the actual DNA sequence.
To enable effective trace back and eliminate the limitation of watermark-based approaches, Kar et al. [13] had proposed a scheme to create digital signatures of DNA molecules in living cells. The main idea is as follows: Take a DNA molecule and sequence it. The result is a string over the alphabet A, C, G, and T, representing the four nucleotide building blocks of DNA. The output of the sequencer is stored in what is called a FASTA file. For interpretability reasons, the FASTA file is annotated by the researcher to create another file called the GenBank file. The authors then use Shamir’s identity-based signature scheme [27], Reed–Solomon error-correction codes [22,23] and the 16 digits Open Researcher and Contributor ID (ORCID –
However, there are significant challenges related to the placement of the signature within the molecule and various types of mutations in more complex molecules that Kar et al. do not address (discussed in more details in Section 2). The current work improves the previous scheme to address these problems (Sections 4 and 5). Moreover, we would like to shorten the size of the signature sequence as much as possible without impacting security. While biologists believe that the size of the DNA has a correlation with its properties within certain bounds, they still do not know by how much a DNA molecule can be expanded without changing the properties of interest. The current work explores other cryptographic algorithms towards this end as well as signature compression schemes (Section 6).
In order to verify a signature, the verifier needs to first identify which part of a signed message is the signature. There are three choices – before the actual message, after it or within it. If the signature is placed within the message, identifying the signature becomes problematic and needs to be addressed during the signature placement time. (The other two cases when the signature is placed before or after the message, are easily addressed by using a counting-length approach.) Unfortunately, when working with DNA molecules, it may not always be possible to place the signature at the end or the beginning of the message. This is because there can be a feature present at these locations. The possible places to place the signature are most likely to be somewhere within the original sequence. For this reason the file that documents the DNA molecule needs to be also accessible to the receiver. Now, these molecule documentation files are created using gene editors that maintain databases of DNA molecule properties. It is conceivable that the databases are updated with location of signature in the molecule. However, these databases may not be consistent across different editors in the sense that receiver’s gene editor may not have all the information about the same set of molecules that the sender’s gene editor has. In addition, there is no universal file format used by gene editors, unlike the FASTA file format.
The document is needed not only to identify and retrieve the signature but also to address the problem of sequence alignment. Thus, we recommend sending the document created by the creator of molecule be also sent to the receiver. This is described in [13]. The updated algorithm described in Section 4 takes into account for any circular or double helix permutations, and the verifiers sequence need not be in perfect alignment with the senders sequence. The purpose of sharing the digital file is not just limited to aligning sequences at the receivers end. The digital file contains descriptions about the physical DNA. There are gene editing software such as Snapgene which can be used to generate documentations or annotations about a sequence. A user can sequence a DNA, provide the FASTA file containing just the raw sequences to the software. The software searches its database for descriptions matching any subsequence within the FASTA file and generates a genbank file which contains the descriptions (from the database) and the original sequence. Refer to Fig. 6 and Fig. 5, the genbank file is generated from the FASTA file, the keyword “ORIGIN” separates the actual sequence and the descriptions. Although the software like Snapgene can generate documentation for some sequences, it cannot describe what is not present in its database. For this, the software have the flexibility of user addition, deletion and update. The user can add more descriptions, can delete wrong descriptions populated by Snapgene, and update a description to include or exclude information.
When a DNA sample is shared, the receiver can only generate the descriptions that the software can generate using its database. The extra descriptions written by the sender cannot be obtained by the receiver unless the digital file is shared. For this reason the digital file also needs to be shared with the receiver along with the physical sample. However, this file, being a separate entity from the DNA molecule, is not strongly tied to the molecule. Sending it separately from the molecule can result in the receiver using a different document file from the one that creator generated. Thus, we propose embedding the document in the DNA molecule itself and sending the whole signed and documented molecule to the receiver. Only this way would the receiver be obtain all the descriptions which the sender intended to share. However, note that placing the document within the molecule brings up the same issue of locating it as the signature. In this work, we develop the necessary techniques to generate, embed, retrieve and validate signatures as well as document in/from the molecule. The details of these procedures are explained in Sections 4, 5 and 8.
We would like to note here that there can sometimes be reasons why the originator may not want to share the entire document file. While the creator may be willing to divulge the properties of the synthesized DNA as a whole, s/he may not be willing to divulge properties of some sub-sequences because of reasons related to protecting intellectual properties or preventing mal-actors from exploiting some of the properties for nefarious purposes. Sending the entire document file may jeopardize these confidentiality needs. However, we do not address these concerns in the current work but is left as a future topic. We assume that if and when the creator is willing to share the document (may be by encrypting parts that cannot be revealed to the receiver) it is embedded in the molecule.
Limitations of earlier work and current contributions
Cyclic shifts and reverse complement
In [13], the signer is required to send the GenBank file along with the physical DNA sample to the receiver. This is because the GenBank file is needed to align the FASTA file (which is the output of a DNA sequencer) in the same order as during the signature generation. Plasmid DNA is cyclic and double-stranded. Following DNA sequencing, any cyclic permutation of the DNA structure is possible. A sequence represented in a FASTA file, and consequently the GenBank file, is thus one of several possible linear representations of a circular structure. For example, in a FASTA file if the sequence was “
Moreover, since DNA is composed of two complementary, anti-parallel strands, a DNA sequencer can read a sample in both the “sense” or “antisense” direction. The sequence may be represented in a FASTA file in either direction. When the sample is sequenced again, the output might be in the other direction, or what is known as the reverse complement. The reverse complement of “
Let us now consider the implications of this characteristic of DNA on the signature generation and verification. The sender has a sequence say “
Let us assume the signature sequence is “
Append the signature after the message: In this case, the sender’s message with the signature embedded looks like – “
Append the signature before the message: In this case, the sender’s message with signature looks like – “
Append the signature between the message: In this case, the sender’s message with signature might look like – “
The problem of recovering the message only occurs when the signature is placed within the message. The other two cases when the signature is placed before or after the message works perfectly fine. However, when working with DNA molecules, it may not always be possible to place the signature at the end or the beginning of the message. This is because there can be a feature present at that location. The possible places to place the signature are most likely to be within the original sequence. For this reason the GenBank file needed to be shared. Only this way would the receiver be able to align the sequence in the same order that the sender had when he signed.
There are several reasons why we may not want to share the GenBank file. The GenBank file is created by the originator of the DNA molecule using a gene editor. Its only purpose is to annotate the DNA sequence. If the DNA is an intellectual property, then the creator of the DNA will be annotating the DNA’s GenBank file with different features of different subsequences of the DNA. While the creator may be willing to divulge the property of the synthesized DNA as a whole, s/he may not be willing to divulge properties of various subsequences. Sending the GenBank file jeopardizes the latter. Moreover, gene editors maintain databases of DNA molecule properties. However, these databases may not be consistent across different editors in the sense that receivers gene editor may not have all the information about the same set of molecules that the sender’s gene editor has. Finally, the GenBank file format is not the only format used by gene editors, unlike the FASTA file format. In order to not share the GenBank file with the receiver, we have changed the signature generation procedure in this work, such that the verification is not dependent on where the signer placed the signature. The details of the new signature generation procedure are explained in Section 4.
Mutations in identifying tags
In our previous work, we defined two identifying tags to demarcate the signature. The start tag was chosen as “
To overcome this limitation, in this work we propose using partial matching techniques such that the start and end tag can be located approximately. This is used in conjunction with error correction codes. Note that since the start and end tags are fixed, we know what we are searching for in the DNA molecule. For example, we may want to look for strings similar to “
Signature length
The length of the signature plays a very important role in this biology domain. Shorter signatures imply less cost of synthesizing the signature into a physical DNA molecule. Shorter signatures will also be less likely to impact the existing functionality and stability of the plasmid during signature embedding. Previously, we used 1024 bit keys and that resulted in 512 base-pair signature. However, 1024 bit keys are no longer considered very strong and not recommended in practice for digital signatures. Generally, 2048 bit keys are used. In our domain, this would result in a 1024 base pair signatures. This length has a higher probability of affecting the characteristics and stability of the plasmid. Furthermore, when synthesizing the signature, presently with a 512 base pair signature the cost is $46.08 – 512 base pairs at $0.09 per base pair. With a 1024 base pair signature, even if the plasmid remains stable and functional, the cost of synthesizing the signature would be $92.16. The new signature scheme with a shorter signature is described in Section 6.
Overview of the DNA signature workflow
We begin by providing a threat model for this work followed by an overview of the DNA signature workflow. Even though the approach is not limited to plasmid DNA, we use “plasmid” to refer to physical DNA molecules that are being created, used or modified in the laboratory. We use the term “sequence” to mean the digital representation of the DNA molecule as a series of nucleotides represented by the letters A, C, G, and T. Note that biologists use the term “sequence” to refer to the DNA molecule or to the data representing the DNA molecule. In the context of this work, we limit the term “sequence” to mean the digital representation to avoid any ambiguity. The documentation, digital signing and verification processes in this work are all conducted on the sequence. The document as well as signature are digital sequences that are converted back to physical molecules and embedded in the DNA molecule.
Threat model
Biological threats: The security of the biotechnology research enterprise depends on trustworthy supply chain transactions [21]. For example, a receiving party may be interested in getting a plasmid or other genetic material from a vendor, a client, or a collaborator. This physical entity will come as few micrograms of white powder or as a few microliters of an aqueous solution. It will be packaged in a small tube with a small label that will provide some information about the tube content possibly in the form of a barcode and sample name. This data points to another source of information such as a product datasheet or a publication for instance. This information is necessary to be able to use the genetic material.
The link between the physical sample and the data describing this sample is very tenuous. Spontaneous mutations can lead to random modifications of the genetic material not reflected on the label or data sheet. Labelling errors can lead to a gross misrepresentation of the tube content. There is no assurance that the information printed on a label will point to a detailed description of the genetic material or that the description of the genetic material will be accurate.
There are a number of common scenarios in which the receiving party may work with genetic material based on misleading information available on the tube label. In a research context, the most likely outcomes of this situation are wastes of time and laboratory processes resulting from experiments that don’t work. In a clinical environment, labelling errors can harm patients and labelling errors are one of the major causes of drug recalls
By encoding identifiers of the sender and the genetic material, this technology makes it possible to verify the authenticity and integrity of the genetic material without relying on the information that may be available on the label.
Cyber threats: The main goal of this work is to protect against the aforementioned biological threats. We do this by using the capabilities of a digital signature and error correction codes. For the cyber part of this work, we assume a polynomial-time adversary, Mallory, who is trying to forge the signature of a reputed synthesized DNA molecule creator, Alice. Alice is trying to protect her IP rights/reputation as she distributes DNA molecules synthesized by her to researcher Ellen. If the attacker, Mallory, is able to forge the signature of Alice then: (a) Mallory can replace the actual DNA created by Alice with her own but keep the signature intact. (b) Mallory can create her own DNA molecule and masquerade as Alice to sign it. (c) Mallory can modify parts of the signed DNA molecule created by Alice. Therefore, for our proposed signature scheme to be secure, we need to show that no polynomial-time adversary can forge a genuine signature without knowing the secret used to sign.
The workflow presented below is enabled by an identity-based signature (IBS) key authority, a trusted entity that creates and distributes keys needed for an identity-based signature scheme. We assume an out-of-band secure channel for such key distribution. We assume that the central authority is secure and trusted by all participants in the system. The description of this central authority is beyond the scope of this article.
Legal threats: There are also a number of legal threats that this approach may potentially be able to protect against. It may limit product liability. A vendor could use this technology to limit representations of functionality to products for which the signature can be validated as opposed to the looser criteria of similarity that are often used in biotechnology. This technology could also be used to help comply with material transfer agreements. Finally, this technology could be used to ensure deniability or facilitate attribution. For example, a research institution such as a government lab may decide to enforce a policy to systematically sign their genetic material. Compliance with this policy could help detect rogue agents who would try to avoid compliance with signature policies. It would also limit the risks of exposure to damaging investigations by law enforcement.
Limits of the technology: The technology does not prevent rogue agents from developing unsigned DNA sequences that may have damaging effects. The situation is similar to digital signature of software. There is no way to force every software developer to sign their codes but digital signature technologies of applications make it possible to develop a trustworthy ecosystem like the Apple App store. Widespread adoption of digital signature technologies limits the risks associated with anonymous applications without completely eliminating it. Similarly, the widespread adoption of digital signatures for DNA sequences would raise the security standards but cannot completely eliminate the inherent risks associated with synthetic DNA molecules.
Recently authors have proposed to use a machine learning approach for attributions of DNA sequences [19]. Digital signatures complement inferential methods by allowing to associate sequences with specific individuals instead of a research group. They also establish the relation between a sequence and an individual with certainty instead of simply providing a likelihood of association. Finally, digital signatures do not require the availability of large datasets. Two parties can securely exchange genetic material without relying a large dataset such as the Addgene repository. However, digital signatures require a minor modification of the genetic material and access to a central authentication authority.
Signature generation and validation workflow
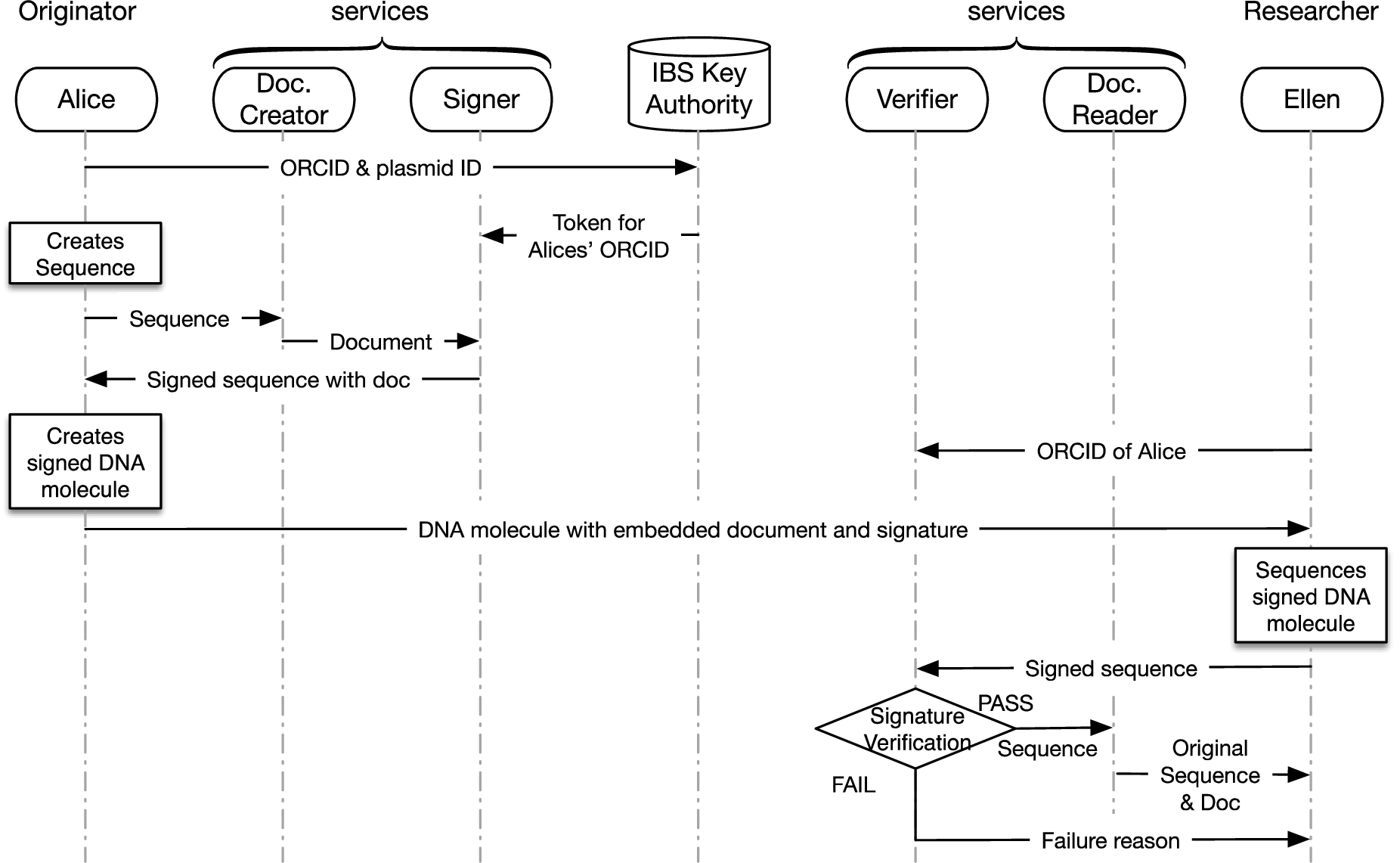
The DNA signature and validation workflow is presented in Fig. 1. There are four major steps in the workflow involving Alice, the Originator, Ellen, the Researcher/User, a Doc creator service, a Signer service, a Doc reader service, and a Verifier service. Note that these services are applications that can either reside on the machines used by Alice and Ellen or can be on remote service providers.
Alice, the Originator, uses a computer software to produce a bioinformatics files that include both the plasmid sequence and its digital documentation. The digital documentation is converted into a DNA sequence by the Doc creator service. Then, the Signer service creates an identity-based digital signature of the sequence that combines the original plasmid sequence and the DNA-version of the documentation.
Ellen, the researcher/user, obtains a plasmid for her use. The plasmid is received via a out-of-band physical communication channel. Ellen uses a DNA sequencer instrument and bioinformatics tools to produce the sequence of the received plasmid.
Ellen imports this sequence into the Verifier service that validates the signature and returns Pass/Fail message. If Fail, the Verifier service also provides explanation of failure.
If the sequence has been validated by the Verifier service, it is passed to the Doc reader service that decrypts the documentation from a sequence and converts it to a standard bioinformatics format.
DNA sign–share–validate workflow.
View of sequenced pUC19 plasmid in SnapGene editor before and after signing.
The steps of the DNA sign–share–verify workflow is shown in Fig. 1. Alice creates a plasmid in her laboratory, submits her ORCID and the plasmid ID to the IBS Authority and gets a corresponding IBS token that is provided to the Signer service. She also creates a sequence for this plasmid (FASTA file) . Next she uses the Doc creator service to create a documentation of the sequence (a GENBANK file). She uses the Signer service, to create a DNA signature sequence that she will add to her design. This sequence is the digital signature and the documentation about the plasmid. It is generated using the signature algorithm described in Section 4 and the documentation algorithm described in Section 8. The digital signature and documentation is then inserted in the sequence between two demarcating sequences used to identify the signature from the rest of the original plasmid’s sequence. We rely on the biological properties of the DNA molecule to determine what is the best position within the molecule where the signature-documentation sequence can be inserted.
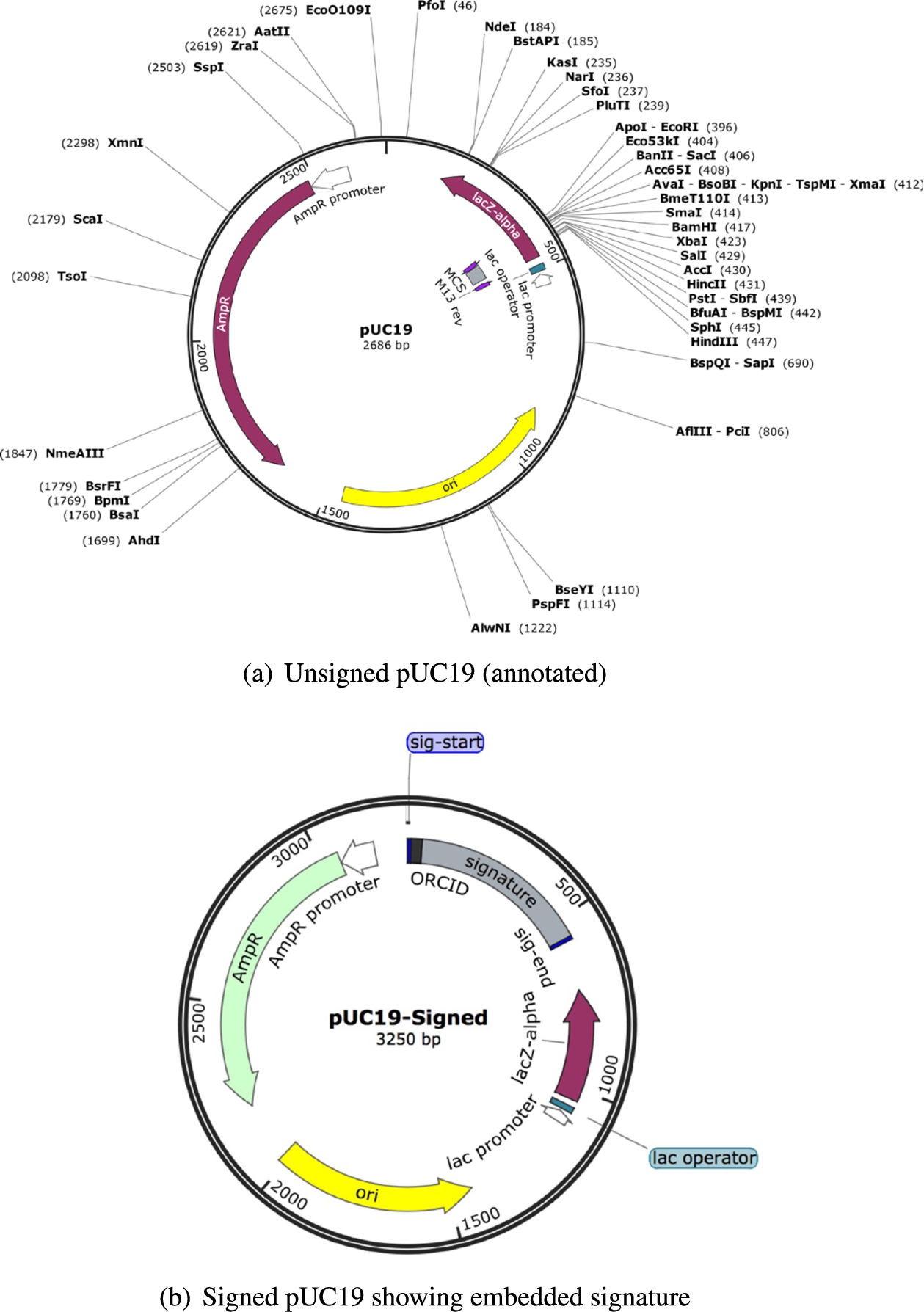
Biologists have observed that there are segments within a DNA molecule that do not contribute to the properties of the molecule that are of interest – the so called “junk DNA”. (In fact, one of the roles that the DNA documentation plays is to identify what are the segments that contribute to the biological properties.) Note that this is not to say that the “junk DNA” does not contribute any properties. It is just that those properties are not of interest to the biologist. The signature-documentation sequence is inserted around such segments. We would like to emphasize two points here: (i) No segment is removed from the original molecule. This means that when the signature-documentation sequence is inserted there is an expansion in the size of the DNA molecule (see Fig. 2). Biologists have experimentally observed that DNA molecules can tolerate some expansion in size without affecting the properties of interest. The tolerable fraction of increase is a function of the size of the original molecule. However, since we do not know the exact fraction of expansion that can be tolerated, a significant focus of this work is in reducing the size of the signature-docmentation sequence. Most of the effort has been spent on experimentally determining what works. We urge the reader to keep this context in mind while reading this paper. (ii) It is possible that some DNA molecule does not allow any expansion in the size. In such cases, no signature-documentation sequence can be inserted and this work is not relevant for such molecules. For the rest of the paper, we assume that we are guaranteed to find a spot within the molecule where the signature-documentation sequence can be inserted. If not, the process exits.
Once the signature-documentation sequence is inserted, the Signer service returns another sequence in the form of a GENBANK file which contains the original plasmid sequence combined with the signature and documentation sequence. Alice then creates a new plasmid (probably by getting the service from a gene synthesis company) corresponding to this signature and documentation sequence. Alice communicates about the original plasmid by using the plasmid ID used to identify the plasmid in the signature and claims ownership of it using her ORCID. When requested she sends the signed plasmid to the User through some physical means (in the biology domain, plasmids are shared. for example, as bio-solutions in test tubes).
Ellen gets the plasmid from some source (may or may not be directly from Alice). Ellen has limited confidence in the plasmid because it came in a hand-labeled tube. So, she decides to get it sequenced completely before doing anything with it. She uploads this sequence of the signed plasmid to the Verifier service to verify the plasmid signature. This uploaded file is a FASTA file. The Verifier service identifies the signature inserted between the two signature tags. The Verifier service will proceed with the signature validation as discussed in Section 4. If the signature is correct, indicated by a PASS response from the Verifier, Ellen will know that the plasmid was signed by Alice and that the physical sequence of the received plasmid corresponds exactly to Alice’s design. Also after successful validation, the Verifier service sends the GENBANK file containing the descriptions and signature that Alice embedded within the plasmid to the Doc reader service, which reveals the original sequence and the corresponding sequence (as allowed by Alice).
Alternatively, the validation service might have determined that the signature was invalid. Several hypotheses could lead to this situation. It is possible that Alice was sloppy and did not manage to assemble the plasmid corresponding to the sequence she had designed. It may have maliciously changed. One could also not rule out the possibility of spontaneous mutations or a labeling error. In this situation, Ellen may decide to proceed with the plasmid based on the similarity of the plasmid sequence and the information available as discussed in Section 5.
For the curious reader we show in Fig. 2 the two sequenced versions of the pUC19 plasmid that we used in our work, both before the plasmid being signed (Fig. 2(a)) and after (Fig. 2(b)). The figures were created via the SnapGene editor that used as input the sequenced versions of both the un-signed and signed molecules. Note in Fig. 2(b), the increased size of the DNA molecule because of the embedded signature.
To present the rest of the work incrementally, we start with the discussion of the core DNA sign–share–validate process. We consider the three major players: (i) The Signer will create the DNA signature and sign a DNA sequence. (ii) The Verifier will use the signature to verify whether the received DNA sequence was sent by the appropriate sender and was unchanged after signing. (iii) A trusted central authority, the IBC key authority, provides the signer with an encrypted token that is associated with the DNA originator’s identity. The token contains the originator’s private key.
Use of error correction in DNA signature: In the digital domain, the digital signature on a message can be used to detect integrity violations. If a violation is detected, the sender can always re-transmit the signed message without incurring much extra cost. However, in the DNA world, we are primarily shipping physical DNA samples. This implies that if a DNA signature identifies that there is an error in the signature validation, then the sample needs to be physically transported and/or synthesized again. This incurs significant cost. DNA mutation is a very natural and common phenomenon. Thus, there is a good likelihood that signature validation will fail. Moreover, associated with the problem of mutation lies the problem of sequencing. When the DNA is processed by an automated DNA sequencer, the output is not always one hundred percent correct. It is dependent on the depth of sequencing, and increased sequencing depth means higher costs. Sequencing a small plasmid to sufficient depth is relatively inexpensive, but for larger sequences, sequencing errors can be an issue. In order to overcome these limitations, we use block-based error correction codes, such as a Reed–Solomon code [23], together with signatures. The presence of error correction codes helps the receiver to locate a limited number of errors (as set by the signer) in the sequenced DNA as well as correct them. The position of the errors and the corrected values are conveyed to the verifier. The verifier can then decide if the errors are in any valuable feature of the DNA or not. If a valuable feature has been corrupted, the verifier can ask for a new shipment, else if the error was in a non-valuable area in the DNA, the verifier can disregard the error and continue to work with it. Generally, there are three types of mutations that occur within a DNA – substitution, insertion and deletion. The error-correction code that is used here only deals with substitution errors.
We now describe our new DNA signature scheme. The steps are shown in Algorithm 1 (for signing) and Algorithm 2 (for verification). To avoid confusion we use the following conventions. The term sample is used to indicate the physical DNA molecule. The term sequence is used to signify the digital counterpart of a DNA molecule. This is generated by sequencing a sample in a DNA sequencer. The raw sequence (output of sequencing) is stored in a FASTA file. The annotated sequence is stored in a GenBank file. The signer creates a physical DNA sample from the signed sequence and sends the sample (only) to the verifier. The verifier sequences this sample to get another sequence that is then verified.
For ease of understanding, we denote the sequence to be signed by the string

DNA signature algorithm accommodating cyclic shifts, reverse complement and mutating tags

New signature verification procedure
Signature generation: The signature generation procedure begins by scanning the GenBank file for the keyword
To account for the problem of placing the signature within the sequence mentioned earlier in Section 2, the signature is generated on the hash of a tweaked version of the sequence. We left rotate a copy of the sequence by
Next, this sequence is passed into the error correction encoder. According to the number of tolerable errors specified by the user, the error correcting parity bits are generated. These parity bits are then converted to some
Signature verification: The verification of the signature embedded in a DNA molecule starts with sequencing the DNA molecule using a DNA sequencer. DNA sequencers produce short reads of 100 to 200 bases of random locations in the DNA molecule. Sequencing reads include errors in which 1 base over 100 to 1000 bases is misread. Since sequencers produce a lot of overlapping sequencing reads, each region of the DNA molecule is read multiple times. Large sets of overlapping reads are processed by specialized software, sequencing assemblers, to produce an assembled sequences corresponding to the consensus sequence. We have provided a more detailed description of the analysis of sequencing data in another paper written for a biology audience [5]. We were pleasantly surprised by the quality of the assembled sequences we were able to produce. The error rate of the assembled sequence is good enough to meet the stringent requirements of digital signature validation. The signature verification procedure applies to the FASTA file produced by the sequencing assembler. It proceeds as described in Algorithm 2.
The receiver sequences the shared DNA using an automated DNA sequencer. The sequence in the FASTA file might not be the in the same order when the sender signed it. That is, after sequencing the shared DNA, the FASTA file may look like –
The first step in the verification procedure is to extract the
Now, it looks for 2
Until this point, we have retrieved
If the FASTA file contains the reverse complement of the sender’s DNA sequence, the entire FASTA file is reverse complemented and then we look for the
The approximate matching technique, shown in Algorithm 3, breaks the entire string in which we are looking for the result into substrings of the length of the input string. Each of the broken substring in the larger string is assigned a score based on how similar it is to the input string. A match is inferred using the highest score. Now in the real DNA, we are looking for sequences of A, C, G, and T. So there might be a case that there are multiple close matches which means that there are multiple starts (or end) tags. In those cases, we use the end tags (or start tags respectively) to narrow our results. The following steps describe how the approximate matching technique works. There can be a total of four scenarios:
Case 1: No mutation in either start or end tags. – In this case, we can find the exact locations of the tags and hence approximate matching techniques are not needed. There can be mutations in any other place which will be handled by the error correction code.
Case 2: Mutation in
Case 3: Mutation in
Case 4: Mutation in both

Approximate matching of tags
Various techniques exist to handle matching of similar strings. These methods measure the distance between strings using a distance equation. One of the most important works in this field is the Levenshtein distance [14]. Other notable algorithms are Damerau–Levenshtein [3,14,30], Optimal String Alignment variant of Damerau–Levenshtein (sometimes called the restricted edit distance) [30], Jaro–Winkler edit distance [11], and Jaccard index [9,10].
We used all these five algorithms for the approximate start and end tag matching. One of the reasons for using all of the above was that we wanted to find out which would be most suited to the DNA domain. For testing, the FASTA file is taken as input and the start and end tag within the FASTA file are manually changed. Next, we search for the location of the defined start and end tags within the mutated FASTA file. The results for each algorithm are summarized on a case by case basis in Fig. 3. As can be seen from the figure, the Jaro algorithm was fairly inaccurate with an average accuracy of only 35.12 %. The Jaccard algorithm fared much better but was still imperfect with an average accuracy of only 95.18 %. All of the three Levenshtein variants were perfectly accurate in their assessment. These results indicate that if accuracy was the chief concern, either of the three Levenshtein variants would be ideal choices.

Accuracy of algorithms per case as a percentage.
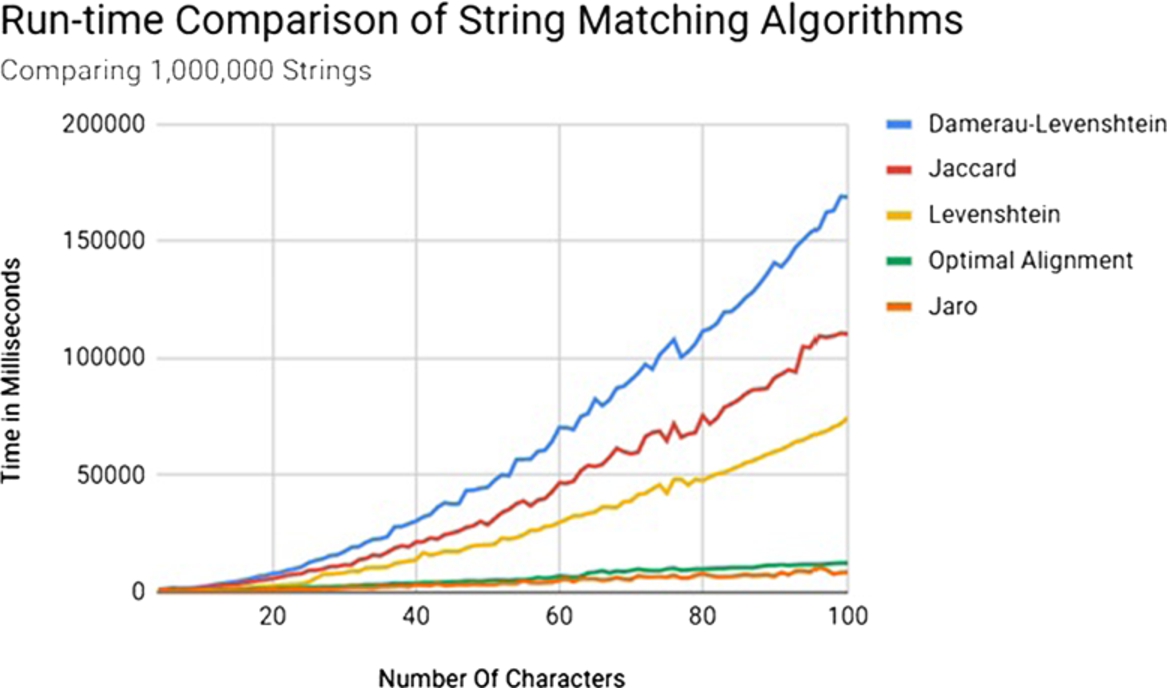
Another important consideration in algorithm selection was speed. While an algorithm may be perfectly accurate in its selection of the closest match to a string it would not help much in practice if the algorithm has an unacceptably long run time. Towards this end, the execution time of the algorithms were also compared. To accomplish this each method was used to compare a series of one million random strings of a set length. A graph of the time in milliseconds (ms) for each algorithm is given in Fig. 4.
Runtime analysis of various algorithms in milliseconds.
As can be seen from Fig. 4, the Jaro–Winkler and Optimal String Alignment algorithms were the quickest, each growing at very slow rates with Jaro–Winkler being slightly faster overall. Taking both of these factors into consideration, we chose the Optimal String Alignment variant of the Damerau–Levenshtein algorithm [30] as our preferred method for string matching.
There are several identity-based digital signature schemes using pairings. Some of the notable schemes are: Sakai–Kasahara [25], Sakai–Ohgishi–Kasahara [26], Paterson [20], Cha–Cheon [2], and Xun Yi [29]. The Sakai–Kasahara scheme described two types of identity-based signatures. One is El-Gamal type and the other is Schnorr type. To identify the most appropriate scheme we first implemented all the above schemes using the Java Pairing Based Cryptography library (jPBC) [4]. We then investigated the signature lengths based on different types of curves that can be used. The time to generate and validate a signature depends on the type of the curve used. We evaluated both aspects: time to sign and verify, and the size of the signature using this algorithm for all the different types of curves present in the jPBC library.
Based on the signature size and the computation cost of signature generation and verification, we identified the best scheme to be the Sakai–Kasahara Schnorr type. We now describe the Sakai–Kasahara Schnorr type identity-based signature scheme. It has four steps: setup, extract, sign and verify.
Setup: The setup generates the curve parameters. The different curves provided in the jPBC library can be used to load the parameters. Let
Extract: Takes as input the curve parameters, the master secret key x, and a user’s identity and returns the users identity-based secret key. This step is performed by the central authority for each user A with identity For an identity Calculate
User A’s secret key is
Sign: To sign a message m, a user A with the curve parameters and the secret key Choose a random Compute Compute Compute
A’s signature for the message m is – (
Verify: The verification procedure is as follows:
Compute
Check
The above equation works because:
Hence,
The signature is a tuple
Signature size using different curves for the Sakai–Kasahara scheme
Signature size using different curves for the Sakai–Kasahara scheme
Based on the signature size, the best performance is provided by the d159, f, and g149 curves. However, the length of the primes are a bit different and also the embedding degree is different. In the d159 curve, the prime is 159 bits and the embedding degree is 6. In the f curve, the prime is 158 bits and the embedding degree is 12. In the g149 curve, the prime is 149 bits and the embedding degree is 10. Keeping in view the small difference in signature sizes and the security related to each type, the better choice is the
The time to generate the signature and verify also depends on the type of the curve because of their properties. Table 2 summarizes the time to sign and verify using the different types of curves.
Average time taken to sign and verify for different types of curves for the Sakai–Kasahara scheme
From the speed perspective, the
Security of scheme: Since we use well-known signature schemes that assume that no polynomial-time adversary can forge a genuine signature without knowing the secret used to sign, it trivially follows that our scheme is also secure.
The Sakai–Kasahara scheme mentioned above generates the shortest signature size among the known identity-based signature schemes. The signature is a tuple (
Now, the elements in the group
Now every elliptic curve and consequently every pairing curve is defined by an equation of the form
This technique can only be applied to signature schemes where the signature contains an element in
In order to recover the Y coordinate during verification we need to do point decompression i.e. retrieve Y using X and the extra byte. The X value is plugged in the equation and the square root modulo prime is calculated. The two values of Y are obtained. Then using the extra byte we know if we should keep the odd Y or the even Y. Note that every curve has a different equation so the decompression method needs to consider the curve and the coefficients a and b. Also, there exists a very easy way to compute the square root modulo prime when the prime modulo 4 equals 3. If prime ≡ 3 mod 4 –
However if prime ≡ 1 mod 4, then there is no one line way to calculate the square root. The
Using these compression methods the signature size which was (32, 40) bytes totalling 72 bytes can be reduced to (20, 21) totalling 41 bytes. Hence the size can be reduced from 288 base pairs (
Self documenting plasmids
In the context of DNA sharing, the receiver gets a physical DNA molecule, which he/she can pass through an automated DNA sequencer and obtain the sequences present within the molecule. The sequence is in the form of a FASTA file which can be used to verify the signature within the molecule. The FASTA file contains just the raw sequences refer to Fig. 5.

Sample fasta (.fasta) file.
However, the receiver has no description about the molecule. Sequence manipulation software such as SnapGene can be used to convert a GENBANK file to a FASTA file and vice versa. When a FASTA file is converted to a GENBANK file, the software searches its database for common annotations. The generated annotations may not be complete or correct every time. Hence, the user has the flexibility to manually add additional annotations that may be required to describe the sample sequence. These manually added annotations are only available to the creator. When the same sample is sent to others, they will sequence it and obtain the FASTA file but the GENBANK file will contain only those annotations that can be automatically generated. In order for the receiver to extract all the feature information for a given sample, the creator would need to share the GENBANK file containing the manually added annotations.
Hence in order to establish a strong tie between the shared GENBANK file and the shared physical sample, the use of dual signature was proposed in [13]. Using a dual signature, it can be verified that the shared GENBANK file containing the descriptions is indeed meant to describe the shared sample and not for any other sample. But having a large set of GENBANK files and a large set of physical samples, it will be difficult to match a sample with its related GENBANK description, i.e. all the samples needs to be tested for the dual signature match.
A sample GENBANK file is shown below in two parts Fig. 6 and Fig. 7. This file corresponds to the FASTA file shown above. The GENBANK file is displayed in two parts due to its length.

Sample GENBANK (.gb) file – part 1.

Sample GENBANK (.gb) file – part 2.
The keyword “ORIGIN” demarcates the annotations/descriptions about the molecule and the actual sequence. The keyword “FEATURES” describes the location of the different features that this sample contains and their location within the sequence. Also it denotes the total number of base pairs in the sample under “source”.
As we are already embedding a digital signature within a molecule, it is also possible to embed the descriptions within it as well. If the descriptions about the molecule can be embedded within itself then there is no need to share the GENBANK file and the physical sample is self documented. The descriptions are text and it can be easily converted to bytes and from bytes to
For the sample GENBANK file, the length of the descriptions (i.e. all text before the keyword “ORIGIN” in Figs 6 and 7) is 3675 bytes. After compression it shrinks to 1493 bytes. Consequently it is a better choice to encode the compressed annotations instead of the actual text. At the time of inserting the annotations, the same challenges we faced for signature insertion and verification because of the circular and double stranded nature of DNA, also appears.
To explain the process, we consider a small example. Let the sample have the following sequence –
Let us assume that when the annotations are compressed and encoded into sequence, it is
When this is shared with the receiver and sequenced it might come out as

A self documenting plasmid generation algorithm

A self documented plasmid reading algorithm
As mentioned earlier, the creator of the DNA molecule may not always be willing to share all information in the DNA document. One simple approach to do this is to encrypt some features of the annotations such that the receiver can only read parts of the annotations. Only the annotations that needs to be read by everyone will be kept in plaintext and encoded as mentioned above. For this approach, there can be several different ways each with its own advantages and disadvantages. Since we are already using identity-based signature, the obvious technique is to use its counterpart identity-based encryption. There are already existing schemes such as Boneh–Franklin IBE scheme [1], Sakai–Kasahara IBE scheme [25] etc., that can be used to accomplish this. However, note that all of the IBE schemes generate the encrypted message as a tuple. The size of the message is doubled. Unfortunately, in this domain we need to keep the message or annotations as short as possible. To what extent we can allow the expansion of a molecule without affecting properties is largely unknown. However, biologists believe that it depends on the size of the original DNA molecule where we would include the annotations. If that sample is large then it is very likely that IBE schemes can be used. One issue with IBE schemes is that it is meant to be decrypted by exactly one particular user. The user’s ORCID which is used to encrypt the annotation will only be able to read the annotations. In cases where we would like to have a selected group of users read an annotation IBE schemes cannot be used.
Another possible method is by using symmetric key encryption like AES. One advantage is that the message size will not expand. But the sender and receiver need to agree on a shared key. This key can be sent to other selected users who will be allowed to read the annotations. But this also implies that a different key is needed for each DNA. Attribute-based encryption techniques is worth exploring in this regard. As of now we have not yet implemented these schemes and is left as a future work.
Conclusion and future work
In this work, we improve the previous DNA signing scheme [13] in several directions. First, we remove the need to share the genbank file by eliminating the requirement of alignment at the sample receiver’s end. The new signature generation procedure is independent of where the signer wants to place the signature. Notwithstanding any cyclic shifts or reverse complements that the receiver may get during sequencing, the signature can still be verified. To account for DNA mutations, we use error correction codes in the signature protocol to correct errors within pre-specified tolerable limits. Our second improvement is a way to locate mutated tags using approximate string matching techniques. This allows us to overcome mutation in the identifying tags and hence we can correctly recover the error correction code. This was a major problem in previous scheme.
Our third improvement is the reduction of signature size. We used pairing based cryptography to improve the previous signature scheme which generated 512 base pair signature to the Sakai–Kasahara scheme which generates 288 base pair signature. Then we further compressed that to 164 base pairs. It will be worth exploring the lattice-based schemes to check if those can be used to generate a shorter signature size than 164 base pairs.
We also explored the possibility of encoding the documentation or annotation about the sample within the sample itself. For just the signature scheme we had removed the need to share any digital file along with the physical sample. But still the receiver after validating the correctness of the sample would have limited knowledge about the description of the sample and the features it has. Previously, we had to share the digital file but included a dual signature such that the receiver can strictly tie one digital file with exactly one physical sample. If we can encode the documentation about the sample also within it, there will be no need to share any extra information and the physical sample will the self validating and self documenting. As of now, the signature and the self documenting parts are implemented as two different modules. In future they will be combined to generate signatures and annotations together.
One of the future directions in this work would involve signing and verifying the same DNA molecule multiple times by different users. Alice signs and sends a DNA sample to Bob and Bob validate Alice’s DNA. Then Bob continues to modify it, then signs it and sends it to Eve. Can Eve only verify Bob’s signature, or is there a way for Eve to track the entire pathway starting from Alice? It would be interesting to see if the concept of aggregate signatures can be applied in these scenarios.
Another direction to be explored is signing a part of the DNA. In this work, we have only applied signatures on plasmids and the entire plasmid sequence. Plasmids are relatively smaller in size than genomes ranging from 2.5 – 25 kilobases, and hence sequencing a plasmid without any errors is feasible. But as we move from plasmids to genomes, the sheer size of the DNA makes it almost impossible to produce and error free sequencing. The error correction code can be used to tolerate some errors but it might not be the optimal solution. It can be a better solution to sign a part of the DNA rather than the entire DNA sequence for example a particular protein sequence. In that way, we can ensure that the protein sequence is unchanged although there might be some errors in rest of the DNA sequence.
Also, it would be interesting to see if we put a signature on top of an existing signature whether the characteristic of the DNA molecule changes or not. If it does not, how many signatures can be inserted before the characteristics of the original DNA molecule begin to change? If we cannot put multiple signatures within the same DNA molecule, how do we remove the signature that was present before signing it again? Does removing the signature also alter the property of the DNA molecule? These are some future directions that we plan to explore further.
We envision an extension of this technology to genomic sequences. Even simple genomes are orders of magnitude larger than the plasmid sequences considered in this article. In addition, their sequence include long repeats and other complex features that are difficult to sequence accurately. As a result, it would not be practical to apply this technique to whole genomes because of the cost of sequencing whole genomes with the high level of accuracy necessary to validate signatures. Instead, we are considering signing genome segments delimited by tags. We could use the tag to amplify these fragments by Polymerase Chain Reaction (PCR – a molecular biology method to rapidly make millions of copies of a DNA molecule) and validate the PCR amplification products. Strain engineers for instance could use this approach to sign the genes they modify in an existing genome.
Footnotes
Acknowledgments
This work was partly supported by the U.S. National Science Foundation’s award #1934573 “EAGER: Development of a tool-chain to write and read self-documenting plasmids”, and award #1832320 “EAGER: Modeling DNA Manufacturing Processes Using Extensible Attribute Grammars”, and by the Colorado State University’s Office of the Vice President for Research Catalyst for Innovative Partnerships Program. The work of Indrajit Ray was performed while serving as Program Director at the U.S. National Science Foundation (NSF) and supported by the foundation’s Independent Research and Development program for staff. Research findings presented here and opinions expressed are solely that of the authors, and in no way reflect the opinion of the NSF, other federal agencies or the Office of the Vice President for Research of Colorado State University.