
Abstract
Background:
Psychotic symptoms of delusions and hallucinations occur in about 5% of persons with Huntington’s disease (HD). The mechanisms underlying these occurrences are unknown, but the same symptoms also occur in schizophrenia, and thus genetic risk factors for schizophrenia may be relevant to the development of psychosis in HD.
Objective:
To investigate the possible role of genes associated with schizophrenia in the occurrence of psychotic symptoms in HD.
Methods:
DNA from subjects with HD and psychosis (HD+P; n = 47), subjects with HD and no psychosis (HD-P; n = 126), and controls (CTLs; n = 207) was genotyped using the Infinium PsychArray-24 v1.1 BeadChip. The allele frequencies of single-nucleotide polymorphisms (SNPs) that were previously associated with schizophrenia and related psychiatric disorders were compared between these groups.
Results:
Of the 30 candidate genes tested, 10 showed an association with psychosis in HD. The majority of these genes, including CTNNA2, DRD2, ERBB4, GRID2, GRIK4, GRM1, NRG1, PCNT, RELN, and SLC1A2, demonstrate network interactions related to glutamate signaling.
Conclusions:
This study suggests genetic associations between several previously identified candidate genes for schizophrenia and the occurrence of psychotic symptoms in HD. These data support the potential role of genes related to glutamate signaling in HD psychosis.
Keywords
INTRODUCTION
Huntington’s disease (HD) is a neurodegenerative brain disease that is inherited in an autosomal dominant Mendelian fashion by way of a CAG trinucleotide repeat expansion in the HTT gene. Patients with HD develop motor symptoms (e.g., chorea), cognitive symptoms (e.g., poor judgment and impaired executive function), and behavioral symptoms (e.g., apathy, agitation, depression, and lack of awareness). In about 4–5% of cases, the behavioral symptoms of HD also include psychosis. Although this represents a small minority of patients, the hallucinations and delusions they tend to experience are often disabling. Indeed, the occasional HD patient who presents with psychotic symptoms but no family history of HD or motor manifestations is sometimes misdiagnosed with schizophrenia. In our previous work, we have reported that psychotic manifestations of HD tend to occur in certain families and not in others [1, 2], thereby suggesting a genetic predisposition to psychosis.
Our observation is consistent with early research in HD genetics, as the presence of genes that modify the clinical features of HD was immediately apparent after the discovery of the HTT gene. The most commonly investigated clinical characteristic in HD is age of onset (AO), and although CAG repeat length accounts for 70% of the variance in HD AO [3], it has been assumed that familial factors other than repeat length account for the remaining variance [4–6]. In the context of our study, we hypothesize that the expression of the psychosis phenotype in HD may depend on the presence of genetic modifiers.
The variability in the expression of psychotic symptoms in HD may be analogous to the variability observed in Alzheimer’s disease (AD), a neurodegenerative disorder where psychosis is common and likely heritable [7]. Barral et al. [8], who compared subjects with late-onset AD and psychotic symptoms to subjects with late-onset AD and no psychotic symptoms, found that several schizophrenia-related genetic variants on chromosome 19q13.12 (ZNF260, ZNF2566, WDR62, and SNX26) were associated with susceptibility to psychosis in AD. This evidence, that genetic factors relevant to schizophrenia are associated with psychosis in other neurodegenerative disorders, motivated us to investigate the possibility of associations between schizophrenia-related genes and the presence of psychosis in HD. This is particularly interesting given that schizophrenia itself may be a polygenic condition resulting from the combined effect of many genes that each have a small effect, as suggested in a recent analysis that identified 108 separate genetic loci potentially contributing to the disorder [9]. Here, we seek to compare the genetic background of HD cases with and without psychotic symptoms to determine whether the genes implicated in other disorders, such as schizophrenia, also contribute to the development of psychosis in HD.
MATERIALS AND METHODS
Subjects with HD (n = 182) were recruited from specialty neurogenetics clinics at the University of Washington (UW) Medical Center and VA Puget Sound Health Care System. These clinics were more likely than community-based geriatric clinics to serve HD patients with neurobehavioral presentations. Thus, although the majority of the HD subjects enrolled in this study had no history of psychosis (HD-P; n = 135), we were able to select 47 HD subjects with symptoms of psychosis (HD+P).
The diagnosis of HD was made on the basis of clinical manifestations, positive family history, genetic testing, and, in some cases, neuropathology. Psychosis was defined by the presence of prominent delusions and/or hallucinations. In 27 of the HD+P subjects, psychotic symptoms were well documented in medical records and were exhibited during direct examination by trained study clinicians; these individuals were classified as meeting a narrow definition of psychosis (or HD+Pn). For the remaining 20 HD+P subjects, psychotic symptoms were very likely to have occurred per informant history but were less well documented in the medical record and were not observed by study clinicians; these individuals were classified as meeting a broad definition of psychosis.
An additional sample of 207 healthy controls (CTLs) were selected from the larger sample of the Consortium on the Genetics of Schizophrenia (COGS) [10]. These CTLs were selected given their lack of both an Axis I diagnosis and a family history of HD, schizophrenia, or other related disorders.
All cases and controls in this study signed informed consent forms approved by the institutional review boards at their respective institutions and were of self-reported European ancestry.
For all subjects, DNA was obtained from peripheral white blood cells and then genotyped using the Infinium PsychArray-24 v1.1 BeadChip from Illumina, Inc. (San Diego, CA). This cost-effective, high-density microarray was developed by the Psychiatric Genomics Consortium (PGC) in collaboration with several leading research institutions for large-scale genetic studies of psychiatric predisposition and risk. The array interrogates about 271,000 verified tag SNPs, 270,000 exome markers, and 50,000 markers that have been associated with schizophrenia, bipolar disorder, autism spectrum disorders, attention deficit hyperactivity disorder, major depressive disorder, obsessive-compulsive disorder, anorexia nervosa, Tourette’s syndrome, and other common psychiatric disorders.
Standard quality control filters were applied to the HD-case and CTL datasets separately using the PGC Ricopili pipeline (https://github.com/Nealelab/ricopili/wiki). Invariant markers and markers with minor allele frequencies <0.01 were removed, as were SNPs with call rates <0.98 or SNPs with Hardy-Weinberg equilibrium p values of <10–10 in HD cases or <10–6 in CTLs. Two HD-P subjects were removed for gender discrepancies, and after comparing ancestry to the HapMap 3 reference panel, 7 HD-P subjects were removed as European ancestry outliers with evidence of Asian, Latino, or African ancestry admixture. Next, the datasets were separately imputed to the 1000 Genomes reference panel [11] using the PGC Ricopili pipeline. Then a final round of quality-control filters were applied to each sample separately, as described above, and the filtered samples were combined to generate a final dataset that included 173 HD cases (126 HD-P cases and 47 HD+P cases, including 27 HD+Pn cases), 207 CTLs, and 5,108,590 variants with a total genotyping rate >0.995. To correct for residual population stratification and genotype pool discrepancies, principal components were calculated on the final sample for use as covariates.
The genes and SNPs for these analyses were derived from three sources and were selected based on availability. First, 18 candidate genes were selected based on evidence of involvement in HD or psychosis in related disorders (e.g., AD, frontotemporal dementia, amyotrophic lateral sclerosis): APC2, C9orf72, CTNNA2, DISC1, DRD2, ERBB4, GRID2, GRIK3, GRIK4, GRM1, HTR2A, NOS1AP, PCNT, RELN, SLC1A2, SMARCAD1, SPTBN2, and ZFPM1 [12–14]. For each of these 18 candidate genes, the total set of directly genotyped SNPs was pruned for linkage disequilibrium, and 159 haplotype tagging SNPs were thus selected for gene coverage.
Second, 43 candidate SNPs were selected based on prior associations with schizophrenia [9] or schizophrenia endophenotypes in the COGS sample [14]. These 43 candidate SNPs derived from 10 genes, including 8 that overlapped with the 18 candidate genes selected above (CTNNA2, ERBB4, GRID2, GRIK3, GRIK4, HTR2A, RELN, and SLC1A2) and 2 that did not overlap with those 18 candidate genes (GRIN2B and NRG1).
Third, 12 candidate SNPs were selected based on evidence of association with age of onset (AO) in HD or related neurodegenerative disorders (e.g., AD, Parkinson’s disease). These 12 SNPs derived from 11 candidate genes (ATG7, CCDC85A, GRIN2B, MAP2K6, MAPT, MTMR10, NPY, RRM2B, VSNL1, ZNF260, and ZNF566), only 1 of which overlapped with the COGS genes selected above (GRIN2B) [15–21].
In total, 214 SNPs were selected for analysis from 30 candidate genes. After accounting for residual linkage disequilibrium, 204 effectively independent SNPs were tested with experiment-wide significant and suggestive thresholds of 0.0002 and 0.005, respectively, as calculated using the Genetic Type I Error Calculator [22].
Association analyses were restricted to variants with a minimum minor allele frequency of 0.05 across the entire sample and were performed using logistic regression with adaptive permutations that assessed empirical significance. To assess genetic factors contributing to psychosis within HD, the primary case-only analyses compared subjects with and without psychosis under both a broad (HD+P vs. HD-P) and narrow (HD+Pn vs. HD-P) definition. In secondary analyses, we separately compared the HD+P, HD+Pn, and HD-P groups to CTLs (i.e., HD+P vs. CTLs, HD+Pn vs. CTLs, and HD-P vs. CTLs), which allowed us to differentiate genetic factors that are unique to the psychotic subtypes of HD. Finally, these results were contrasted with those comparing HD subjects without psychosis to controls (HD-P vs. CTL), which would presumably identify variants associated with HD in general. The complete set of results across all SNPs and group comparisons is provided in Supplementary Table 1.
RESULTS
The characteristics of the subjects, including CTLs, are shown in Table 1. The 126 non-psychotic HD (HD-P) subjects, including 77 males and 49 females who were of an average age of 55, did not differ significantly in gender or age from the 47 psychotic HD (HD+P) subjects, including 32 males and 15 females who were of an average age of 48.5. In contrast, the CTLs were more likely to be female and younger. The higher number of males in the HD groups is unsurprising given that this sample was partly ascertained from a VA hospital population (https://www.va.gov/vetdata/veteran_population.asp), whereas the CTLs were ascertained from a community sample.
Sample description
Given that many of these HD subjects were recruited to participate in research prior to the identification of the HD gene, only a subgroup of these subjects have CAG repeats available. Of the HD-P subjects, 77 had a clinical diagnosis of HD confirmed by molecular genetic studies, with a mean CAG repeat expansion size of 44. All 47 HD+P subjects had HD confirmed molecularly, with a mean CAG repeat size of 45. The two groups did not differ significantly by CAG repeat size. Although CTLs did not undergo CAG repeat testing, detailed family histories were obtained in these subjects, and a family history of HD would have excluded the subjects from participating in the original COGS study.
As shown in Table 2, we identified 25 SNPs that were specifically associated with either HD+P or HD+Pn. These SNPs are found in the following 10 candidate genes: CTNNA2, DRD2, ERBB4, GRID2, GRIK4, GRM1, NRG1, PCNT, RELN, and SLC1A2. See the Supplementary Table for additional analysis. We performed a pathway analysis of these genes, and Fig. 1 shows that interactions have been observed for most of the associated genes and that most of these interactions are involved in regulating glutamate receptor signaling.
Association results
Odds ratios are listed only for SNPs that are associated with psychosis and that have no evidence of an association with HD-P. Odds ratios in bold are significant at p < 0.05, with all others having trending p values <0.10. *Indicates p values <0.01. **Indicates p values <0.005 and that meet at least suggestive levels of significance experiment-wide. Non-significant associations are listed as “ns.”
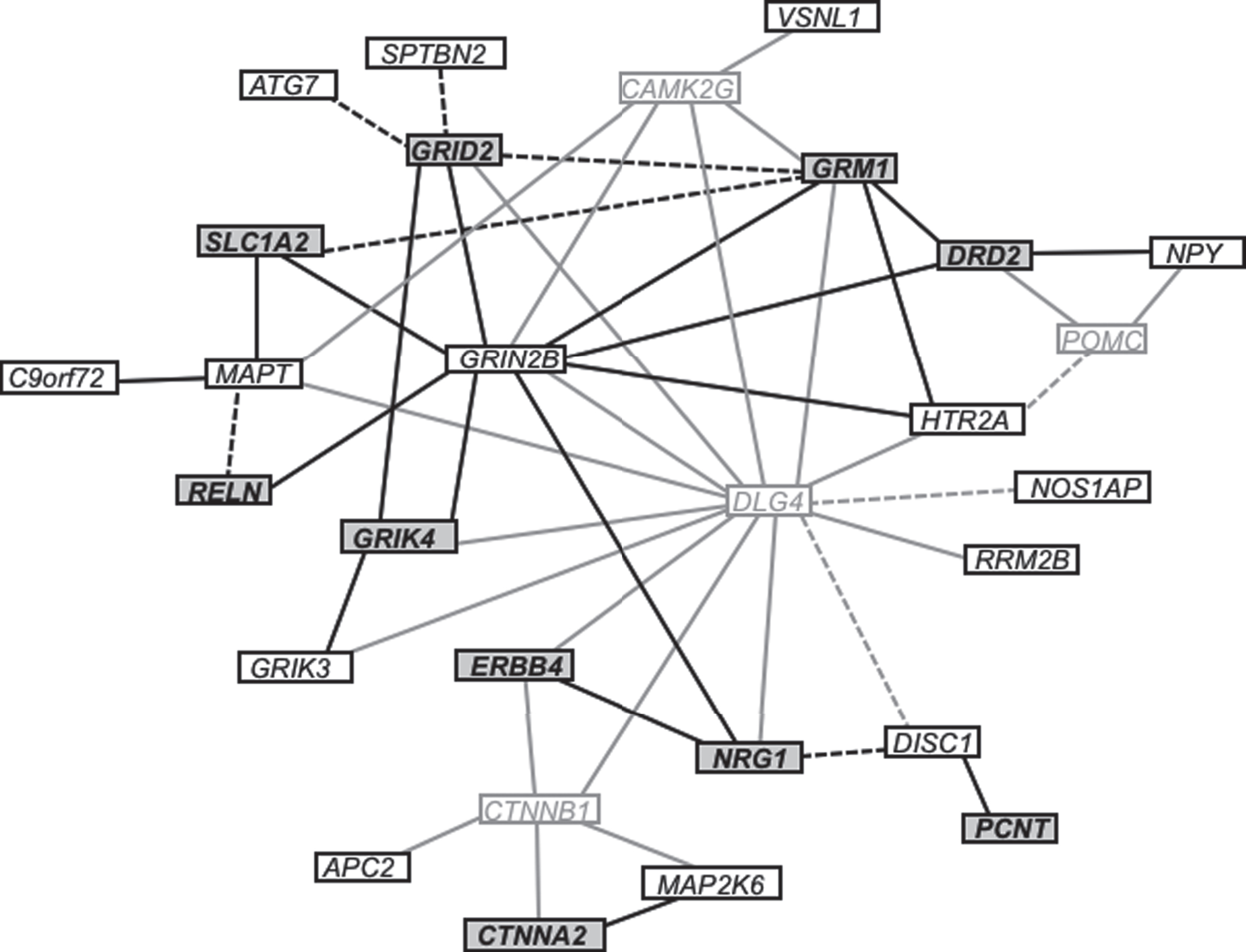
Gene interaction network illustrating the relationships among investigated genes. Connections between genes indicate that the respective proteins jointly contribute to a shared function, though not necessarily to a shared physical interaction. Genes depicted in bold text with a dark gray background were specifically associated with psychosis in HD. Genes depicted in black text with a white background were investigated but not found to be associated with psychosis in HD. Genes in light gray text with a white background were not tested for association but form key connections between investigated genes. Solid lines represent known interactions, whereas dashed lines represent predicted interactions based on co-expression or text mining.
DISCUSSION
This is the first study to directly investigate the genetics of psychosis in HD. The candidate genes identified here are particularly noteworthy given their apparent involvement in the regulation of glutamate receptor signaling. Glutamate interacts with both ionotropic and metabotropic glutamate receptors (e.g., NMDA and mGluRs, respectively), and it has been postulated to play an important pathogenic role in the excitotoxic neuronal cell loss that takes place in HD [23]. Our findings also parallel past studies that have described specific molecular pathways and modifier genes that may contribute to the phenotypic expression of HD.
Genetic association studies have shown that SNPs in GRIN2B (rs890, rs1806201) have modifier effects on AO in HD [24]. These findings are particularly interesting in the context of our study because a recent European HD Network REGISTRY investigation [25] found that the association of rs1806201 with HD AO was only evident when the HD cohort was divided according to the type of clinical presentation of HD. In this case, the inclusion of the GRIN2B genotypes in the model increased the R2 more substantially in the AO of the 241 subjects who presented with psychiatric symptoms (specific types of psychiatric symptoms not reported) than the AO of the 538 subjects who presented with motor symptoms. Although this increase was modest and none of these SNPs have been demonstrated to have functional effects, the finding broadly affirms that modifying genes likely play roles in the phenotypic expression of a single-gene Mendelian disorder like HD.
Although none of the variants in GRIN2B that were tested here, including the two variants discussed above, were significantly associated with psychosis in HD, several of the genes that have been shown to interact with GRIN2B (e.g., GRID2, GRIK4, GRM1, DRD2, NRG1) demonstrated genetic associations with psychosis in HD, suggesting that they may jointly contribute to a shared function. Indeed, as seen in Fig. 1, we observed a number of interrelated functions and interactions between the candidate genes for HD-related psychosis in this study. Likewise, many of these genes have been previously associated with schizophrenia. In two independent samples, 8 of the 10 genes associated with HD-related psychosis in this study (CTNNA2, ERBB4, GRID2, GRIK4, GRM1, NRG1, RELN, and SLC1A2) have shown pleiotropic association with 3 or more schizophrenia endophenotypes, primarily endophenotypes related to cognitive dysfunction [26–29].
Of the 10 genes associated with psychosis in HD, NRG1 has been the most frequently studied in relation to schizophrenia [26–29]. Neuregulin-1 is a trophic factor that signals through the activation of the ErbB receptor tyrosine kinases, such as ErbB4, which plays a crucial role in neurodevelopment and in the modulation of NMDA receptor signaling, processes often disturbed in psychosis [30–32]. Since Steffansson et al. [27, 33] first identified NRG1 as a candidate gene for schizophrenia, many studies have confirmed the finding, including animal studies demonstrating an overlap in the NRG1 behavioral phenotype (as well as its receptor, ERBB4, and the NMDA receptors) and schizophrenia. There have also been some negative studies related to NRG1 and psychosis, but Gardner et al. have argued that population stratification and geographic NRG1 haplotype clustering may account for these negative findings [34]. Other studies have shown a disproportionate disruption of genes in the neuregulin and glutamate pathways in psychosis [35, 36], and Go et al. [19] have extended the association of NRG1 to Alzheimer’s disease with psychosis (ADP), demonstrating that variation in NRG1 may increase the risk of developing hallucinations and/or delusions in the context of the neurodegenerative processes of late-onset AD.
Given that a disturbance in glutamatergic transmission has been suggested in the pathophysiology of schizophrenia [37, 38], our discovery of an association between HD+P and the SLC1A2 gene is also notable. SLC1A2 (solute carrier family 1, member 2) encodes one of the major glutamate transporters, EAAT2 (excitatory amino acid transporter 2). Mutations in mouse SLC1A2 have been shown to impair glutamate uptake, and the resulting excess glutamate leads to subsequent excitotoxicity. In humans, there have been both positive [39] and negative [40] genetic association studies of schizophrenia conducted on SNPs in SLC1A2 in Chinese and Japanese populations, including a study in which Zhang et al. [39] found a significant genetic association with a SNP in the SLC1A2 gene in a Han Chinese population with 2,128 cases of schizophrenia and 3,865 controls. Likewise, Afshari et al. [41] have reported an association between a novel mutation in SLC1A2 and schizophrenia in a large Palauan kindred, and several of the SLC1A2 SNPs tested in our study have been associated with cognitive impairment in schizophrenia. In related psychiatric disorders, Medina et al. [42] found downregulation of mRNA transcripts of SLC1A2 in the hippocampal brain tissue of 13 subjects with major depressive disorder compared to 10 healthy controls, and de novo point mutations in SLC1A2 have been shown to cause epileptic encephalopathies and neurodegenerative disorders, including a SNP in SLC1A2 that has been associated with Parkinson’s disease in a Chinese population [43]. Given these past findings, it is interesting to discover that there is also evidence regarding possible glutamatergic involvement in the pathophysiology of HD.
Although CTNNA2, GRID2, PCNT, and RELN have received less attention than NRG1, ERBB4, or SLC1A2 in relation to schizophrenia, these genes may harbor de novo variants that play a role in schizophrenia and related disorders [13, 44]. Finally, D RD2, the primary target of effective antipsychotics, recently demonstrated genome-wide significance in a large collaborative association study of schizophrenia, as did several genes involved in glutamate signaling [9].
Both cortical and subcortical brain regions are affected in HD by a marked initial loss of dopamine-innervated GABAergic striatal cells [45]. This striatal degeneration disrupts the activation of the cortico-subcortical pathways that provide the majority of the excitatory glutamatergic input into the striatum. However, because the clinical consequences of striatal degeneration on these pathways varies from patient to patient, this degenerative process may play an important role in the development of the variable phenotypes in HD [46]. Specifically, for some patients, these alterations in striatal medium-sized spiny neurons may increase the sensitivity of NMDA receptors in a subpopulation of neurons, and this increased sensitivity may result in the expression of the psychosis phenotype in HD. These complex mechanisms underlying receptor regulation of glutamatergic release are beyond the scope of our analyses, but it is likely that genetic variants (along with environmental factors) may influence these mechanisms, their neurophysiological responses, and the underlying pathogenesis of HD. Increasing our understanding of these complex relationships may thus offer important new insights in our understanding of inter-individual differences in HD.
The largest caveat of the present study is its small sample size, which limits the power to identify true associations, particularly for the narrow definition of psychosis. Although no single SNP reached an experiment-wide significance level of 0.0002, several SNPs were experiment-wide suggestive with p values <0.005, despite the small sample. We also confirmed the association with psychosis in HD of many candidate genes and SNPs previously associated with schizophrenia. Importantly, these are likely not spurious findings, as the associations were specific to psychosis in HD and not shared with non-psychotic HD subjects, suggesting that the identified genetic risk factors may modify the expression of psychosis in HD.
Future studies may also be useful in better characterizing the relationships between gender, ethnicity, and psychosis in HD. Although we found no differences in gender and psychosis within HD, our CTL group was not matched for gender, and it is possible that gender-specific effects may exist in genes (both in the subjects and in the parents who transmitted the genes to the subjects) that modify the psychosis phenotype in HD. We also opted to focus exclusively on individuals of European heritage, as we lacked a sufficient sample size to account for ethnicity-based differences in genetic heterogeneity. This means that our results are only directly generalizable to individuals of European ancestry. Larger sample sizes of HD subjects with well-characterized psychotic phenotypes are thus necessary to confirm these findings and explore potential gender and ethnicity effects. However, given the variability in the definition of psychosis across HD studies, insufficient sample sizes currently exist to conduct more informative studies.
Finally, environmental factors likely play a role in modifying the development of psychosis in HD, as most phenotypes are likely the result of interactions between genetic and environmental exposures. Valuable insights into the complex molecular events that drive HD and psychosis pathology will emerge as we learn to better dissect these complicated genetic and environmental relationships. Molecular genetic studies are also necessary to elucidate the mechanisms by which these 10 genes may exert their biological effect in the development of psychosis in HD. Such studies may yield critical information toward the development of effective treatments for psychotic symptoms in HD.
CONFLICTS OF INTEREST
We have no conflicts of interest to report.
Footnotes
ACKNOWLEDGMENTS
This work was supported by Merit Review Award #I01 BX002241 from the US Department of Veterans Affairs (DVA) Biomedical Laboratory Research and Development Service and by the Geriatric Research, Education, and Clinical Center at the VA Puget Sound Health Care System. The contents do not represent the views of the US DVA or the US Government.