
Abstract
Amongst the main reasons people at risk for Huntington’s disease (HD) have for undergoing predictive genetic testing are planning a family and prevention of passing on an expanded CAG-repeat to future offspring. After having received an unfavourable test result, a couple may consider prenatal testing in the foetus or preimplantation genetic diagnostic testing (PGD) in embryos. Testing of the foetus or embryos is possible by means of direct testing of the expanded repeat. Optimal reliability in testing the foetus or embryos requires the establishment of the origin of the repeats of both parents in the foetus. For PGD the analysis is combined with or sometimes solely based on identification of the at-risk haplotype in the embryo. This policy implies that in the context of direct testing, the healthy partner’s CAG repeat lengths in the HD gene are also tested, but with the expectation that the repeat lengths of the partner are within the normal range, with the proviso that the partner’s pedigree is free of clinically confirmed HD. However, recent studies have shown that the expanded repeat has been observed more often in the general population than previously estimated. Moreover, we have unexpectedly observed an expanded repeat in the non-HD partner in four cases which had far-reaching consequences. Hence, we propose that in the context of reproductive genetic counselling, prior to a planned pregnancy, and irrespective of the outcome of the predictive test in the HD-partner, the non-HD partner should also be given the option of being tested on the expanded allele. International recommendations for predictive testing for HD should be adjusted.
Keywords
INTRODUCTION
Huntington’s disease (HD) is a hereditary late onset neurodegenerative disease marked by involuntary movements, cognitive deterioration, and behavioural disturbances with onset for the majority of patients between 30 and 50 years of age [1]. So far, symptomatic treatment is available that might reduce the signs of HD and improve the well-being of the patient. No treatment is yet available that affects its etiology [2, 3]. HD is caused by a pathogenic mutation of the HTT gene involving CAG trinucleotide repeats that exceed 35 [1, 4]. Identification of this mutation enabled individuals at risk to find relief from anxiety and uncertainty. Since, predictive genetic testing is offered following internationally endorsed recommendations [5].
Planning a family and prevention of passing on an expanded CAG-repeat to future offspring are amongst the main reasons for individuals at risk for Huntington’s disease (HD) to apply for predictive testing [6]. Identification of the expanded allele in one of the partners enables a couple to consider prenatal testing (PD) when pregnant, or preimplantation genetic diagnosis (PGD) before an existing pregnancy. Recent studies have shown that the frequency of expanded repeats (ranging from intermediate to full penetrance alleles) in the general population is higher than expected [7]. Indeed, in our out-patient HD predictive and prenatal testing clinic we have observed expanded repeats in partners without an HD family history. Consequently, we argue that irrespective of the test outcome of an individual at risk for HD, the partner should also be offered testing if family planning is a major and compelling reason to have a predictive test. This test should preferably be done before the couple is actually stepping into PD or PGD.
In this paper we describe four cases in which the couples and clinicians involved were confronted with an unexpected outcome of prenatal testing. In all cases expanded repeats were observed in (or stemming from) the presumed non-HD side of the couple.
THE CAG TRINUCLEOTIDE REPEAT
Four categories of repeat size ranges in the HTT gene have been identified [8]. First, 40 CAG repeats or more are full penetrance alleles (FPAs) and will cause symptoms. Larger repeat sizes are associated with an earlier age at onset [9, 10]. Second, 36 to 39 repeats are incomplete or reduced penetrance alleles (RPAs) and may cause symptoms, usually later in adult life. Third, 27 to 35 repeats represent the category of intermediate alleles (IA), which are not associated with symptomatic disease, but sporadically show instability and have the potential to expand into the RPA or FPA range within one or more generations, mainly through the paternal line [8, 11]. Fourth, repeats of 26 or less are not associated with HD and are stably transmitted to the offspring.
Reports about the extent of the instability of IAs and RPAs show differences in HD-populations. In the Venezuelan HD kindreds, out of 69 transmitted alleles in the IA range, only one expansion from 33 to 35 CAGs was found, but none expanded into the RPA or FPA range while 14% of transmitted RPAs expanded into the full penetrance range [12]. In a Portuguese study, instability in FPAs was found in 66% of all transmissions [13, 14]. Semaka et al. (2010) examined the intergenerational stability of IAs. Overall 30% of the alleles were unstable upon transmission with more expansions than contractions. The mean change in the CAG size was 1.39 but the expansion range was from +1 to +23 repeats. Expansions were present in 24% of the transmissions from the 31 to 35 CAG range and in 14% of the transmissions from the 27 to 30 CAG range [15, 16].
The repeat has been found especially unstable when paternally transmitted [17]. Moreover, the larger the expansion of the CAG repeat, the higher the risk for further expansion [2, 18]. However, it has been demonstrated by analysing the CAG repeat size in single spermatozoa that IAs can expand to RPAs and FPAs [16, 19].
THE CAG TRINUCLEOTIDE REPEAT IN THE GENERAL POPULATION
IAs in the HTT gene have been observed in 1–7% of the general population, which can be considered as relatively commonly present [11, 20]. In British Columbia, confirmed (0.7%) or probable (7.1%) new mutations were found in HD patients with a negative family history [21]. Kay et al. (2016) reported that 1 in 400 individuals from the general population has a repeat >35 (=0.246%), of which 0.041% has a repeat >39. General population RPA penetrance rates are lower than penetrance rates extrapolated from clinical cohorts [7]. In a Dutch study on 1,690 patients tested to confirm or exclude HD, 46,9% had a repeat size less than 27. Sixty patients (3.6%) had an IA of whom half had no family history concerning HD [22].
PRENATAL DIAGNOSIS AND PREIMPLANTATION GENETIC DIAGNOSIS
To prevent passing on the CAG expansion to their offspring, individuals carrying an expanded CAG repeat have the option of prenatal diagnosis (PD) or PGD.
PD involves DNA testing of chorionic villi, obtained by transcervical or transabdominal biopsy in the 10–13th week of pregnancy or of amniotic fluid cells, obtained by amniocentesis from the 15th week of pregnancy on.
PGD is performed on cleavage stage embryos on the third day after fertilisation with ICSI or on blastocyst cells after trophectoderm biopsy on day 5/6. For HD, embryos with two normal alleles and/or normal haplotypes are transferred to the uterus [23, 24]. Usually linkage methods such as STR marker analysis or karyomapping will be added to or will replace analysis of the expanded and normal alleles to optimize accuracy.
Direct testing of the expanded repeat and/or testing with markers closely linked to the HD locus enables testing of the fetal/embryonic material [25, 26]. Maternal samples are required in case of prenatal diagnosis to exclude maternal cell contamination. Identification of the origin of the repeats or haplotypes of both parents in the fetus or embryo is needed to secure optimal reliability in PD as well as in PGD. This policy implies that in the context of direct testing, the healthy partner’s CAG repeat lengths in the HD gene are also tested.
PD and PGD for HD are not as frequently chosen as one would think based on the number of at-risk individuals [27, 28], but for a substantial number of couples morally compelling because they don’t want to pass the risk on to their future children. Some couples might decide to only have children if they receive favourable test results themselves. They intend to refrain from having children in case of unfavourable results and consider it as unacceptable confronting their children with a parent who becomes affected by HD in the future. Others with FPAs opt for prenatal testing with the consequence to have the pregnancy terminated in case of unfavourable results, or for PGD.
CLINICAL OBSERVATIONS
We present four cases in which expanded CAG repeats were unexpectedly observed in partners without a known HD family history at the time of counselling and genetic testing (see Fig. 1 and Table 1).
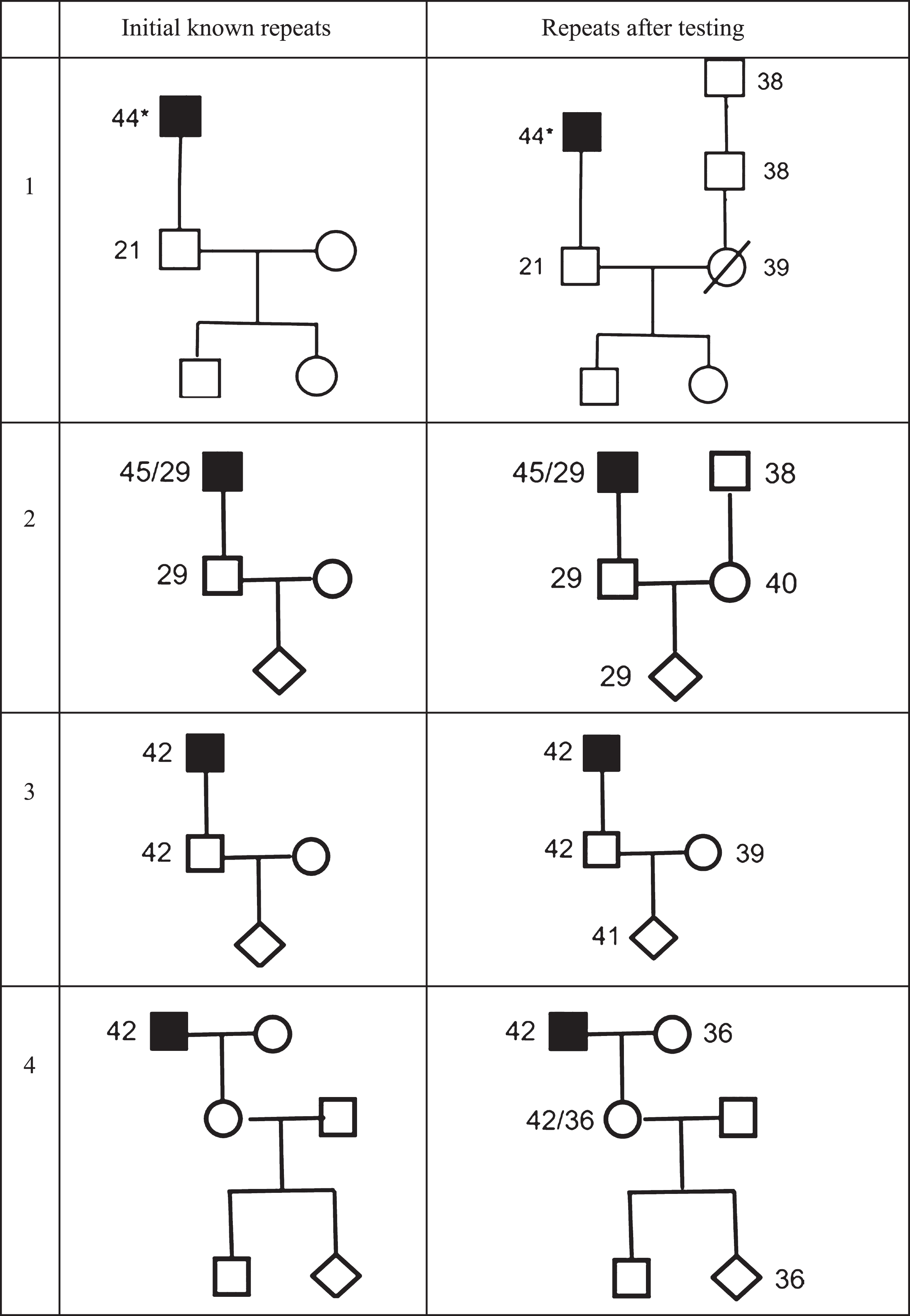
Four couples at risk for Huntington’s disease applying for prenatal genetic testing. *highest number of repeats presented.
Initial and actual risks in 4 couples applying for prenatal testing for Huntington’s disease
Case 1
Twenty years ago, a married man at 50% risk and 33 years of age, requested testing for reasons of planning a family. He didn’t wish to pass the expanded allele on to the next generation and didn’t want to confront children with a father suffering from HD. HD was not known in his wife’s family. The couple intended not to have children if he should be identified as carrier of the expanded allele. He received a favourable test result (21/23) and two children were born. Four years ago, his wife died from a brain tumour. Recently a relative informed him about information received at another genetic centre that HD had been diagnosed in his wife’s family and that his wife’s father and grandfather were identified as having an RPA. He wished to have his wife’s DNA analysed to inform his adolescent children. His wife proved to have an RPA (39). His daughters are consequently at 50% risk of having an expanded allele (most likely an RPA, but as further expansion cannot be excluded, possibly also an FPA).
Case 2
A man (31 years) at 50% risk applied for predictive testing. He was found to have 29/17 CAG repeats which was still a great shock. He interpreted the result as less bad than an FPA, but he and his wife still felt they should as much as reasonably possible avoid exposing their future offspring to the risk of repeat expansion and related decisional issues. When his wife became pregnant, they therefore insisted on prenatal testing. After extensive counselling the couple could accept termination of the pregnancy on the basis of 32 repeats. In the context of PD, the woman, from a presumed non-HD family, was tested and proved to have 40 CAG repeats. Genetic analysis of her parents showed that her father has a reduced penetrance allele of 39 repeats. The woman reported that her father showed changes in his personality and cognitive decline since his early sixties which was explained by alcohol abuse.
Case 3
A man (24 years, carrier HD, 42/23 repeats) and partner (23 years) wished to not pass on the HD to future children and applied for prenatal testing in their first pregnancy. For analysis and interpretation of results the alleles of both partners were examined. The results showed 41 repeats in the foetus but, unexpectedly, also 39 in the mother. No family history of HD could be found in the mother’s ancestry, but analyses showed 39 CAG repeats in mother’s healthy mother.
Case 4
When she was pregnant, a woman (26 years) learned that her father was clinically diagnosed with HD which was confirmed with genetic testing (42 repeats). Her mother had died a few years earlier from cancer. Six months after delivery she requested predictive testing because she considered another pregnancy but only after she was informed about her genetic status. The test result was 42 and 36 repeats, implying that her mother must have been carrier of an RPA but also that her first child has no escape of being at increased risk. When she became pregnant, she applied for prenatal testing and considered termination only if the foetus would carry the full penetrance allele. The couple applied for PGD for this reason but were refused. Because any foetus would carry at least an RPA, transfer of an affected embryo was considered unacceptable from a medical ethical perspective. In this case we see the consequences of the ignorance of the woman’s parents about their double risk to transfer an expanded CAG allele.
DISCUSSION
Couples present for genetic counselling and predictive testing for reasons of planning a family with the strong wish to exclude passing HD on to the next generation [27]. Genetic counselling implies that the couple is comprehensively informed which enables them to make an informed decision. Comprehensively informed implies that they are informed on all possible test outcomes of predictive testing (full expansion, reduced penetrance, intermediate and normal alleles) and on reproductive options (PD and PGD) [5]. Couples should also be informed on new insights from research and clinical experience, with the inclusion of new insights into the dynamics of (expanded) repeats. Finally, couples who wish to exclude HD in future offspring should be acknowledgeable of population risks of HD, which is the core message of this paper.
According to the international recommendations concerning reproductive options, preconception counselling should be available to couples where one partner is at risk of HD or is a carrier of the HD gene expansion (rec 7.0.1) [5]. It is also recommended that the importance of preconception counselling is stressed, because of the timeframe in making a decision about testing during an on-going pregnancy. Moreover, such preparation may help to prevent a couple at 50% risk first requesting counselling and testing at the time of a pregnancy, which is a very stressful situation due to the limited time available and the potential for consecutive adverse outcomes, i.e. identification of expanded CAG in the foetus informing the partner at risk that he/she carries the expanded repeat (double bad news).
On the basis of the high frequency of intermediate alleles in the general population and our clinical experience, all cases of prenatal testing have a possibility of ascertaining an IA that was inherited from the non-HD side of the couple, implying that there are also risks of expanded CAG-repeats for the non-HD partner [14, 29–31]. Consequently, all couples considering prenatal testing or PGD for HD may benefit from a discussion on the possibility of unexpected results that may have uncertain clinical implications [31]. How couples come to understand and interpret an intermediate allele result need to be further explored. Individuals may have difficulties to grasp the clinical implications of intermediate alleles for themselves and relatives. Especially in families without a generations-long history of HD, understanding and interpreting intermediate alleles may be a caveat [32].
These new insights inevitably mean that with the present policy of testing the non-HD partner in the context of PD, double bad news should also be reckoned with a possible result of such testing. For PGD the possibility that both partners are carriers entails a greater chance of the couple being written left with no embryos that are free of expanded repeats. According to the international recommendations only embryos with two non-HD alleles should be transferred (rec 7.2.5). Moreover, as presently the non-HD partner will only be tested when the HD partner is found to be at risk, this policy may lead to a false sense of security about future offspring. If partner testing depends on the result for the HD partner, then if the latter is found to have a normal CAG repeat, prenatal testing or PGD is no longer appropriate. Consequently, with the present policy an expanded CAG repeat in the non-HD partner will not be timely identified. Our cases illustrate these implications of the new data on population risks.
In case 1, the relief after learning the favourable result in the man enabled the couple to start a family free from HD. However, learning that the partner did have an HD background was devastating. In retrospect testing the partner would have been justified to exclude any risk to pass the expanded allele on to their future children. The children have to face now their 50% risk to have inherited the RPA, knowing that the expansion risk for RPAs is considerable [12]. Testing a partner and finding an expanded allele implies that he/she might inform the extended family with all ramifications. Being a messenger of bad news is an additional strain. However, knowing (for offspring) outweighs the not-knowing of family members.
Although PD for an individual with an intermediate allele of the HD gene (case 2) is justified (rec 7.1.4), it is not recommended to terminate a pregnancy on the basis of an intermediate allele result (rec 7.1.8) [5). Following the recommendations, the clinical geneticist discussed the risk of expansion of a 29 IA. Expansion of a 29 IA into the HD-range has been reported once 33] but the clinical diagnosis was debated [22, 34]. The couple regarded the unexpected outcome of a 40 FPA in the non-HD partner as a justification of prenatal testing.
Also, for identified carriers of the expanded FPA, such as in case 3, pre-conceptional testing of the non-HD partner might have been an acceptable option in hindsight. The woman in case 4 learned about her father’s diagnosis when she was pregnant of her first child. At that time, she and the unborn child were at 50% and 25% risk respectively.
The couples in cases 2, 3 and 4 have received double bad news which had to be dealt with. Moreover, the unexpected information on the expanded allele in the non-HD partner (case 2 and 3) or in the non-HD parent (case 4) might lead to reconsideration of the reproductive decisions the couple had initially made. All couples felt responsible toward their future offspring and made use of prenatal testing or would have used it (case 1). The risk on an RPA or FPA in the unborn proved in all cases higher than was previously thought: 50% in case 1 and 2, 75% in case 3, and 100% in case 4.
Had the couples been informed on the additional risks, they might all have decided differently. In our view, this illustrates the importance of making couples who wish to avoid transmitting HD to their offspring aware of those additional risks and to do so as part of preconception counselling, to allow them to not only opt for testing the non-HD partner preconceptionally, but also for doing so irrespective of the testing result of the HD partner.
Some may argue that offering such testing to the non-HD partner amounts to a form of population screening in unsuspected individuals without a positive family history. Moreover, given that HD is a serious non-treatable late onset disorder, the testing offer would be at odds with internationally accepted criteria for responsible population screening [35]. Although we agree that population screening for expanded CAG repeats would be problematic in view of the high likelihood of an unfavourable harm to benefits ratio for those tested, we do not think that the testing offer under consideration would indeed fall under the heading of population screening. The point is that in the context of counselling couples at risk of passing on HD to their offspring, the additional information that testing the non-HD partner may yield is directly relevant for dealing with the couple’s request for medical help. This not only holds for the present policy where partner testing is already done with an eye to the reliability of PD or PGD, but also for preconception testing of the non-HD partner independently of the results of the HD partner. Given that the counselees present as a couple with a shared wish to exclude the HD partner’s transmission risk, it seems reasonable to assume that they would also want to be informed about a hitherto unknown additional risk for that same outcome in the non-HD partner, as that - as we have shown — may affect the choices open to them.
No doubt questions arise regarding the benefits and drawbacks of providing couples with the option of testing both partners preconceptionally. In addition to allowing for reproductive decision making on the basis of more comprehensive information while avoiding double bad news during pregnancy, the benefits of such a test include reassurance of the absence of an expanded allele in the non-at risk partner in a family already burdened by HD. The possible adverse consequences of the test include anxiety raised by unfavourable outcomes and the need to inform relatives as detection of an expanded allele may have far-reaching consequences for personal health and reproductive choices. Moreover, the offer of testing the non-HD partner may be felt to add to the burdens of an already emotionally charged situation. Couples have often spent years of extensive counselling not only to explore reproductive options, but also to anticipate emotional responses, and face the impact on their relationship. Adding new far-reaching information as a result of testing the non-HD partner may potentially change the dynamics of dealing with the relevant risks as a couple in ways that are difficult to predict. This is a further reason for making sure that such testing is done preconceptionally rather than under time pressure in the context of a PD or PGD procedure, but also for carefully evaluating the psychosocial impact of the change of policy that this would entail. On those conditions, however, we feel that couples wanting to avoid the passing on of HD to their offspring should indeed be made aware of the population risk and be offered the option of testing the non-HD partner accordingly. Consequently, we suggest adjustments of the recommendations for predictive testing for HD, as well as for prenatal testing and PGD [5].
Another question that arises is whether offering preconception testing of the non-at-risk partner should also be considered for couples at risk of transmitting other dominant genetic disorders with or without treatment options. Here again, we have the opinion that this may well be justified in the light of the often strongly held wish of these couples to avoid passing on the condition to their offspring. However, this needs further exploration that is beyond the scope of this paper. In each case, and regardless of the precise condition, the bottom line remains that the ultimate decision is taken after extensive genetic counselling in which all pros and cons are considered.
Conclusion
We conclude that couples at risk for HD who apply for predictive testing because of reproductive decision-making should be informed about the population risk of having an expanded CAG repeat. In addition, partners of individuals at risk for HD should get access to testing CAG-repeats, preferably before a planned pregnancy or PGD procedure and irrespective of test result of the HD partner. Recommendations for predictive genetic testing for HD need to be adjusted accordingly. We also recommend that the proposed policy should be carefully evaluated.
CONFLICT OF INTEREST
The authors report no conflict of interest.