
Abstract
Background:
Selective serotonin reuptake inhibitors (SSRIs) target the serotonin transporter (SERT) and are commonly prescribed for depression in Huntington’s disease (HD) patients. However, SERT expression in HD has not been carefully evaluated in patients or mouse models.
Objective:
In this study, we investigated SERT levels in HD patients and HD mouse models.
Methods:
We obtained HD patient brain striatal samples and matched controls, as well as brain tissue from CAG140 and R6/2 mice. SERT mRNA and protein levels were analyzed using quantitative RT-PCR and immunoblotting.
Results and Conclusions:
We found that SERT protein, but not mRNA is markedly increased in grade 4 HD patient striatal tissue. These findings suggest posttranscriptional or translational SERT dysregulation as a possible etiologic factor modulating psychopathology in HD. Interestingly, SERT expression is variable in mouse models of the disease. Increased SERT levels are demonstrated in the brain of CAG140 mice, a full-length knock-in mouse model of the disease, but not in the striatum of the R6/2 fragment murine model of the disease. Based on this parameter, the CAG140 huntingtin knock-in mouse model is more suitable than the R6/2 model for the study of serotonergic pathway pathology in Huntington’s disease.
INTRODUCTION
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease caused by expansion of CAG repeats in the huntingtin gene, encoding a polyglutamine stretch in the N-terminus of the protein (HTT) [1–3]. Repeat length of 35 or fewer polyglutamines in the resulting protein is normal, and repeat length beyond 40 is pathogenic [4]. HD symptoms include progressively disordered motor function, weight loss, and emotional disturbances including depression [5]. The HD neuropathological classification system has 5 grades (0–4), which is based on severity of striatal atrophy under macroscopic examination and the severity of striatal neuronal loss and degree of reactive astrogliosis under microscopic examination [6].
Serotonin (5-HT), a monoamine neurotransmitter, is a key regulator of mood and is associated with cognitive function [7, 8]. The serotonin transporter (SERT) is responsible for serotonin reuptake from the synapse into the synaptic boutons [8, 9]. Thus, SERT plays an important role in regulating 5-HT signaling. SERT immunostaining intensity increased in the striatum of HD patients compared with controls [10]. Selective serotonin re-uptake inhibitors (SSRIs), commonly used to treat depression, inhibit SERT function as noncompetitive antagonists to decrease transporter affinity for serotonin [11, 12]. As a result, transport through SERT decreases resulting in increased synaptic 5-HT levels [13]. SSRIs can not only alleviate the psychiatric symptoms in HD patients [14], but also delay neurodegeneration via increased neurogenesis [15]. In addition, SSRIs extend the survival of N171-82Q and R6/1 transgenic HD mice [16, 17].
A number of genetic HD murine models are used to study the disease [18–22], including fragment and full-length HTT transgenic mice as well as knock-in mice [20, 21]. Each model has advantages and disadvantages with respect to experimental modeling of the disease depending on the mechanistic focus of the study [20]. However, the suitability of using a mouse model to investigate SERT expression in HD has not been evaluated. Since no mouse model replicates all aspects of human disease pathology, we undertook a study to find a murine model with relevant SERT pathology.
MATERIALS AND METHODS
Human brain tissue
Postmortem striatal tissue (SBB6) and globus pallidus (SBB7) specimens were generously provided by the New York Brain Bank at Columbia University. They were processed as described [23]. Standard brain blocks (SBB) 6 and 7 were used for this study (see below). SBB6.1/6.2 are caudate, putamen, and nucleus accumbens. SBB7/7.1 is globus pallidus [23]. Specimen details are described in Table 1.
Human brain tissue information
Mouse models
R6/2 mice, which carry the promoter sequence and exon 1 of a mutant human HTT gene with approximately 150 CAG repeats, were obtained from JAX (Bar Harbor, ME). A colony was maintained by breeding R6/2 males with B6CBAF1 females (JAX). CAG repeat length was determined for every mouse in the colony and we used mice with 150 + /- 10 CAG repeats. PCR genotyping was performed [24]. Eight- and eleven-week-old R6/2 mice were used in the experiments. Brain tissue from CAG140 mice were generously provided by Dr. Michael Levine (UCLA) [25]. In CAG140 mice, the Htt exon 1 is replaced with the human HTT exon 1 sequence with 140 CAG repeats. Whole brain homogenates from 11-month-old CAG140 mice were also used in the experiments. All live vertebrate experiments were performed in compliance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pittsburgh.
Antibodies
The following primary antibodies were used: human serotonin transporter [26] (cat#ST51-2, Mabtechnology, GA, USA) 1 : 1000 dilution; mouse serotonin transporter [27] (cat#HTT-GP-Af1400, Frontier Institute, Japan) 1 : 1000 dilution; β-actin (Sigma, A5316) 1 : 2000 dilution; synaptophysin (cat#S5768, Sigma & cat#ab32127, Abcam,) 1 : 5000 dilution. The following secondary antibodies were also used at 1 : 10,000 dilution: goat anti-mouse IgG (IRDye 800CW, Li-Cor, Lincoln, NE) donkey anti-guinea pig IgG (IRDye 800CW, Li-Cor, Lincoln, NE); goat anti-rabbit IgG (IRDye 680CW, Li-Cor, Lincoln, NE).
Immunoblot
Brain tissues were homogenized and lysed in 1x RIPA buffer (Millipore #20-188) with 1% protease inhibitor cocktail (Sigma, P8340) by 21 G and 23 G needles in sequence, and supernatant was collected after centrifuging at 13000 g for 15 minutes. Protein concentration was measured by Bradford assay (Bio-Rad, CA, USA). Samples (30 μg protein) of each sample were run in 10% SDS-PAGE gels and transferred to a PVDF membrane (Immobilon-FL 0.45 μm, Millipore, Bedford, MA, USA). Membranes were incubated in blocking buffer (SEA BLOCK, ThermoFisher Scientific) for one hour at room temperature, then probed with primary antibodies at 4°C overnight. The membranes were then washed three times with 10 ml PBST (0.1% TWEEN-20 in 1x phosphate buffered saline (PBS) buffer) for 10 minutes, and then probed with secondary antibodies for one hour at room temperature. After three 10 minute washes with PBST, the membranes were scanned by Odyssey CLx Imaging System (Li-Cor). The signal was measured using the image studio software (Li-Cor). The mouse doublet SERT immunoblot bands were previously verified as specific using SERT-KO mice [27]. Quantified SERT signal was normalized for equal protein loading using β-actin.
Quantitative real-time RT-PCR (qPCR)
Brain tissues were homogenized in Trizol (Invitrogen) using 18 G, 23 G, and 25 G needles in sequence. Total RNA was isolated by following the protocol using a RNeasy Mini Kit (QIAGEN). 1000 ng total RNA was reacted in 20 μl volume high-capacity cDNA reverse transcription mixture (ThermoFisher), at 25°C for 10 minutes, at 37°C for 120 minutes, at 85°C for 5 minutes and ending at 4°C (Bio-Rad, T100 Thermal Cycler). TaqMan gene expression assays were applied for 100 ng cDNA products in universal PCR master mix (TaqMan), and the primers are SERT (human, Hs00984349_m1), β-actin (human, Hs99999903_m1), SERT (mouse, Mm00439391_m1) and β-actin (mouse, Mm00607939_s1). Thermocycling conditions were as follow: 50°C for two minutes for UNG optimal, followed by 95°C for 10 minutes for polymerase activation, 40 cycles at 95°C for 15 seconds and anneal at 60°C for one minute (Bio-Rad, CFX96™ Real-Time PCR Detection System). Real-time results were quantified by the ΔΔCt method [28] and normalized with β-actin.
Statistical analysis
Statistical significance was evaluated in Prism 7 (Graphpad Software) by using t-test for two groups and ANOVA followed by Dunnett’s test for multiple comparisons. Each experiment has at least three biological repeats, and p < 0.05 was taken as statistically significant.
RESULTS
SERT protein is increased in human HD striatum
The neuropathological severity of HD is classified into 5 grades, from grade 0 to grade 4 according to the severity of damage [6]. The neurodegenerative process commences and most severely affects the striatum, affecting other brain regions as the disease progresses. To determine whether SERT is dysregulated in HD striatum, we compared SERT levels in three grade 2 and four grade 4 HD samples with four controls (Table 1) by qPCR and immunoblotting.
We evaluated mRNA levels in human striatum tissues from HD patients and control subjects using qPCR. We found no significant differences in SERT mRNA levels between control and HD samples (Fig. 1A). Membrane SERT levels known to be regulated post-translationally via constitutive transporter internalization followed by degradation in the endosomal and lysosomal pathways [29]. We therefore examined SERT protein levels by immunoblot. After normalizing with β-actin, we found that SERT protein levels increase in the striatum of grade 4 HD patients (Fig. 1B-D). Striatal neurons project to globus pallidus and SERT proteins are presynaptically localized, as indicated by increased binding of a SERT ligand to globus pallidus as compared with striatum [30]. Thus, we also examined the SERT protein levels in globus pallidus and found no significant difference between control and grade 2-4 HD patients (Fig. 1F, G). These data suggest that SERT protein synthesis in the soma is increased in HD, but this increase is not translated to increased SERT in the projections.
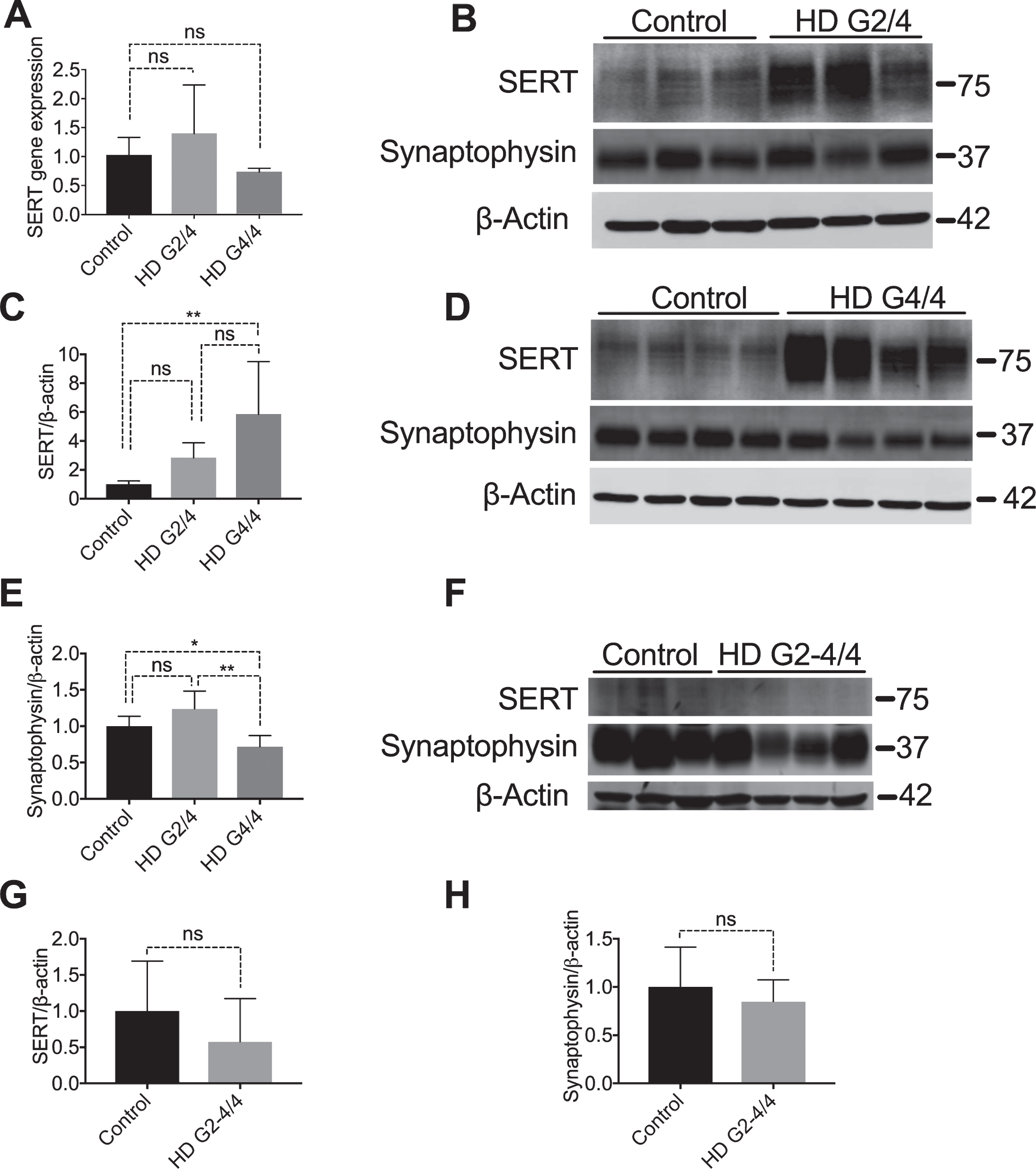
SERT protein levels are increased in human HD striatum. (A) Total mRNA was isolated from control (n = 3), HD grade 2 (n = 3) and HD grade 4 (n = 3) striatal brain sections and SERT mRNA was quantified by qPCR. Data shown are after normalization to β-actin by the 2-ΔΔCt method, and data were analyzed using ANOVA followed by Dunnett’s post-test for multiple comparisons, graphs show means±SD of n = 3. (B) Striatal lysates from non-neurologic controls (n = 3) and HD grade 2 patients (n = 3) and (D) HD grade 4 patients (N = 4) were immunoblotted for SERT and synaptophysin. (C) SERT protein expression and (E) synaptophysin protein expression were quantified for each blot by normalizing to control after normalized to β-actin, band intensity differences among control, HD G2/4 and HD G4/4 were analyzed by one way ANOVA for both SERT and synaptophysin (C, E). (F) Globus Pallidus from non-neurologic control individuals (n = 3) and HD grades 2 to 4 patients (one grade 2, two grade 3, and one grade 4; n = 4) were immunoblotted for SERT and synaptophysin. SERT (G) and synaptophysin (H) were quantified by normalizing to β-actin. After normalizing to β-actin, band intensity differences among control and HD G2-4/4 were analyzed by t-test for both SERT and synaptophysin (G and H). (G2/4 = Grade 2, G3/4 = Grade 3, G4/4 = Grade 4). Graphs show means±SD, *p < 0.05; **p < 0.01; ns p≥0.05.
Consistent with known synapse loss observed in HD pathophysiology [31, 32], synaptophysin, a synaptic marker, was decreased in grade 4 but not in grade 2 HD striatal samples (Fig. 1B, D, E), although this increase was not reflected in the globus pallidus (Fig. 1F, H). Since SERT levels are high even when there are fewer synapses present, the increased SERT protein level cannot be explained by an increased number of synapses in the protein lysate. Therefore, the striatal increase in SERT protein, but not mRNA, suggests that post-translational mechanisms are responsible for the increased striatal SERT protein levels.
SERT protein level is decreased in R6/2 mice
R6/2 mice, one of the most commonly used fragment genetic murine models of HD, demonstrate rapid disease progression with brain atrophy beginning at four weeks of age, and symptomatic disease onset detected at approximately seven weeks. An early event identified at three weeks of age is decreased synaptosomal mitochondrial protein import, suggesting early synaptic pathology [24]. Progressively through their life, R6/2 mice have decreased neostriatal volume, striatal neuron atrophy, and increased astrogliosis, with a concomitant reduction in striatal neurons, as well as activation of caspase cell death pathways [33, 34]. The lifespan of these mice is approximately 14 weeks [35]. To examine SERT levels across the lifespan of these mice, we obtained brain mRNA and brain protein lysates at 8 and 11 weeks of age.
We analyzed SERT mRNA in R6/2 striatal tissue using qPCR and compared these results to the HD patient data. At early/mid disease stage (8 weeks), there is no change in SERT mRNA levels (Fig. 2A), similar to the human grade 2 data (Fig. 1A). However, in advanced disease in the R6/2 model (11 weeks), SERT mRNA levels in R6/2 striatum are significantly increased compared to WT (Fig. 2A), which differs from the advanced HD patient data (Fig. 1A, no change in SERT mRNA levels).
Because we found that SERT protein levels increased in HD grade 4 patient striatum, we measured SERT protein levels in striatal as well as cortical lysates in the R6/2 mice at 8 and 11 weeks of age. In contrast with human patient data (Fig. 1C, D), SERT protein levels were decreased significantly in both striatal (Fig. 2) and cortical (Fig. 3) lysates of 8-week-old (Fig. 2B, D; Fig. 3A, C) and 11-week-old (Fig. 2 C, D; Fig. 3B, C) as compared to wild type. We also measured synaptophysin to determine if the loss of SERT protein in the lysates corrected with the loss of synapses; however, it did not (Fig. 2B, C, E; Fig. 3A, B, D).

SERT protein is decreased in 11-week-old R6/2 mice striatum. (A) Total mRNA was isolated from 8- and 11-week-old mouse striatum, WT (n = 5), R6/2 (n = 5)/at each age point. SERT mRNA was quantified by qPCR. Data shown are after normalization to β-actin by the ΔΔCt method. (B-D) Striatal lysates from 8- (B) and 11-week-old (C) WT (n = 5) and R6/2 (n = 5) at each age group were immunoblotted and quantified for SERT (D) and synaptophysin (E) by normalizing to control after normalized to β-actin. Graphs show means±SD of n = 5, *p < 0.05; **p < 0.01; ***p < 0.001; ns p > 0.05 using one-way ANOVA to compare WT vs R6/2 at the indicated ages.

SERT protein is decreased in 11-week-old R6/2 mice cortex. (A-D) Cortex lysates from 8- (A) and 11-week-old (B) WT (n = 5) and R6/2 (n = 5)/at each age group were immunoblotted and quantified for SERT (C) and Synaptophysin (D) by normalizing to control after normalized to β-actin. Graphs show means±SD of n = 5, *p < 0.05; ns p > 0.05 using one-way ANOVA to compare WT vs R6/2 at the indicated ages.
Like the human data, the qPCR and immunoblot data demonstrate different results, suggesting that in both cases SERT protein levels are post-transcriptionally modulated. However, SERT protein levels in R6/2 mice were opposite to that of grade 2 and 4 human striatum HD protein levels, suggesting that R6/2 mice do not replicate the striatal SERT accumulation seen in human HD.
SERT protein level is increased in CAG140 mice
CAG140 mice were developed by replacing mouse Htt exon 1 with human HTT exon 1 sequence with 140 CAG repeats, leading to expression of a full length mouse/human chimeric mutant HTT protein [20]. Thus, CAG140 mice are genetically closer to human HD as compared with R6/2 mice. CAG140 mice show hyperactivity at 4 weeks, gait abnormalities at 48 weeks and progressive gliosis, and decreased striatal projection neurons at 12 months. CAG140 mice have a normal lifespan [36]. Since our data shown above suggest that SERT is regulated post-transcriptionally, and due to having limited samples, we analyzed protein levels but not mRNA levels in CAG140 mice. We evaluated SERT levels in whole brain lysates from 11-month-old CAG140 mice, and found that SERT protein was increased in CAG140 mice as compared with wild type mice (Fig. 4A, B). Synaptophysin levels were similar in control and CAG140 mice (Fig. 4A, C). These results suggest that CAG140 mice may be physiologically more relevant as a model to investigate regulation of SERT expression in HD.
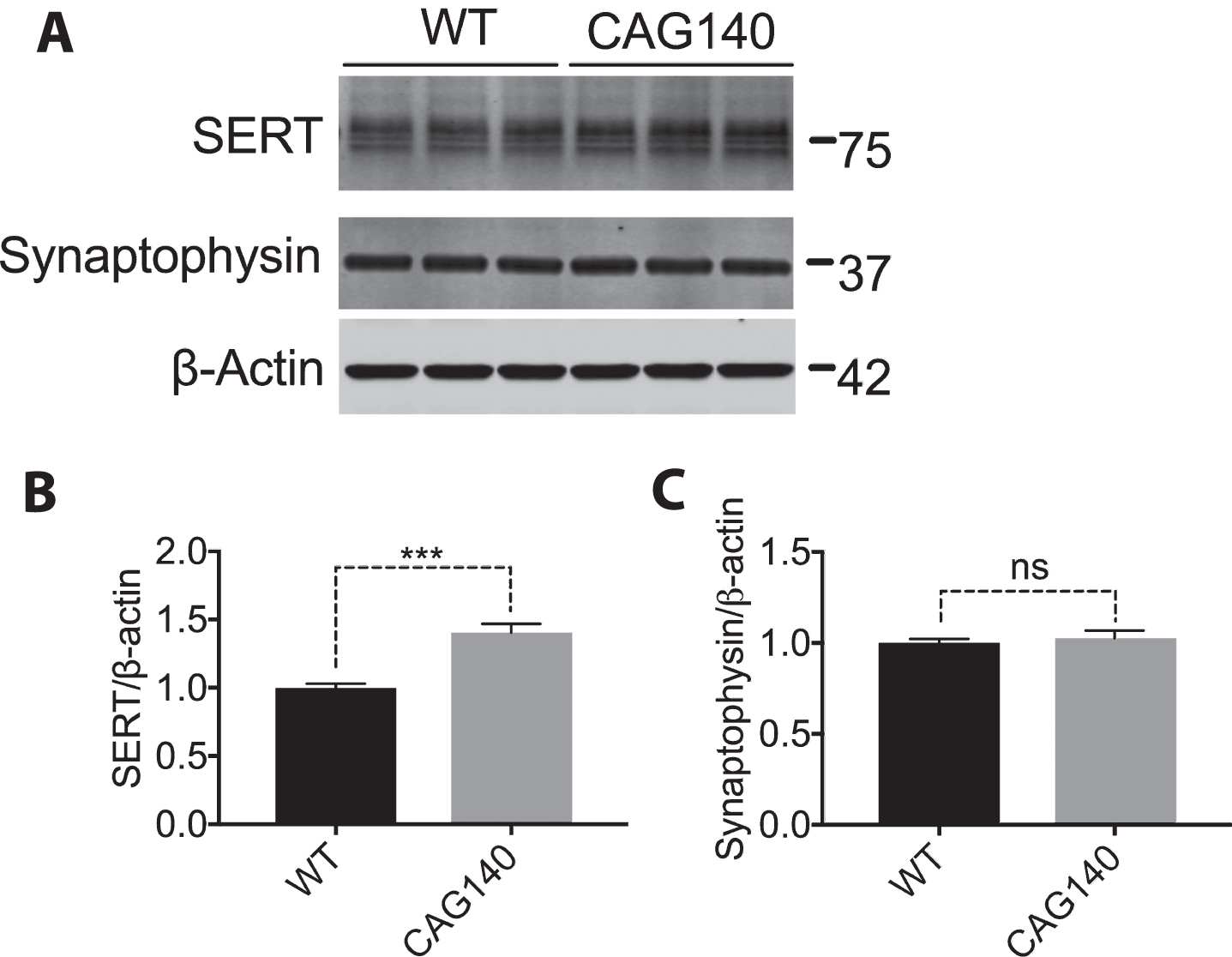
SERT is increased in the brain of 11-month-old CAG140 mice. (A, B, C) Whole brain lysates from 11-month-old WT (n = 3) and CAG140 (n = 3) were immunoblotted for SERT/Synaptophysin and quantified by normalizing to β-actin. Analysis was done to compare WT to CAG140 using a t-test. Graph shows mean±SD of n = 3, ***p < 0.001.
DISCUSSION
Using HD patient striatal tissue, we demonstrate that the serotonin transporter SERT protein levels are significantly increased in grade 4 HD patient striatum when quantified by immunoblot. This is consistent with a previous genome-wide study that showed that SERT mRNA expression levels were increased in the striatum of HD patients [37] and that by using semiquantitative immunohistochemistry, SERT is increased in grade 4 patient striatum [10]. In this publication, the apparent increase of SERT staining in HD was attributed to brain atrophy [10]. Our data are consistent with respect to the increase in SERT protein level, but the decrease in synaptic markers combined with equal total protein loading suggests that this is caused by changes in SERT regulation in HD. Furthermore, our data demonstrate that the increase in protein is seen only in the striatum, and not the globus pallidus.
There are several possible mechanisms that could lead to increased SERT levels in the striatum, but not the globus pallidus, of HD patients. Recent studies have shown that SERT is primarily degraded through late endosomal and lysosomal degradative pathways [29]. Lysosomes have been reported to be dysfunctional in HD patient brain tissue [38]. Together with our data, this could suggest that dysfunctional lysosomal activity in HD may lead to an accumulation of SERT in striatal neuronal cell bodies. Alternatively, it is possible that SERT protein accumulates in the neuronal cell body, represented by the striatum, and is not effectively transported to the projections (globus pallidus). SERT protein trafficking from endoplasmic reticulum to the plasma membrane subdomains could be affected by many mechanisms including the binding of phosphoatidylinositol-4,5-biphosphate (PIP2) to SERT proteins [39], truncated C-terminus of SERT proteins [40], binding of various proteins to the C-terminus or other regions of SERT proteins [41, 42]. Further study is required to clarify what is the root cause of SERT protein accumulation in the striatum. Regardless of the cause of the increase in striatal SERT protein, dysregulated SERT protein levels could lead to pathologically dysregulated synaptic 5-HT, altering 5-HT signaling and ultimately affecting neuronal survival. Future studies to investigate the cause of increased striatal SERT as well as the downstream effects of increased SERT protein expression may lead to insights into HD pathophysiology.
Given the heterogeneity of genetic design between mouse HD models, it is critical to understand what aspects of the pathophysiology they mimic and which they do not [43]. Fragment genetic murine models of human HD including the R6/2, R6/1, and N171-82Q mice express the N-terminal exon of human mutant Huntingtin. These mice have rapid disease progression, including shortened lifespan, weight loss, motor performance abnormalities, and neuropathological sequelae [20, 21]. Murine huntingtin knock-in mice are a more accurate genetic model of HD, in which an expanded CAG repeat is inserted into the murine huntingtin homologue, including the CAG140 and Hdh/Q72–80 mice. These mice have mild chronic disease progression and neuropathological phenotype compared with fragment models and have a normal life span that does not model human disease progression [20]. When choosing an experimental model, it is important to examine the degree to which each model mimics the endpoint pathophysiologic event being evaluated. In comparing models for serotonin regulation, we find that CAG140 mice mimic the dysfunction in SERT that is seen in HD patients. R6/2 mice, on the other hand, have decreased striatal SERT protein levels instead of the increased levels found in HD patients.
It is of interest to note that given the recently identified role of mitochondria in melatonin synthesis [44], and the fact that serotonin is a key intermediate of melatonin synthesis; alterations in the biosynthesis pathway and receptor mediated events of these two ligands (melatonin and serotonin) may play a role in mitochondrial and synaptic dysfunction in HD. This will be a subject of future investigation.
Synaptophysin is a marker of neuronal presynaptic vesicles. We examined synaptophysin in HD patient striatum and globus pallidus, 8- and 11-week old R6/2 striatum and cortex, and whole brain lysates from CAG140 mouse. We found that synaptophysin levels were decreased in human HD grade 4 samples but not in 11-week old R6/2 striatal and in CAG140 whole brain lysates. Thus, synaptophysin protein expression from late stage R6/2 striatum does not recapitulate HD patient grade 4 pathology, although our data from 8-week old R6/2 confirms a previous report suggesting that synaptophysin was decreased in R6/2 mouse striatum [45].
In summary, our findings show that HD patients have significantly increased SERT protein levels in the striatum but not the globus pallidus. The downstream effects of this alteration in 5-HT pathway in HD is unknown, but likely has an impact into the prevalent mood disorder in HD. Furthermore, the CAG140 murine huntingtin homologue knock-in mouse may be a suitable model for SERT study in HD while the R6/2 transgenic fragment human HD murine model is not. There is no mouse model that completely recapitulates human HD. Each mouse model has its strengths and weaknesses and it is therefore important to use complementary models and always relate to known findings in human tissue.
CONFLICTS OF INTEREST
Robert Friedlander serves as the Chief Medical Officer of NeuBase Therapeutics. Diane Carlisle serves as a scientific consultant for NeuBase Therapeutics. No funding from Neubase Therapeutics was received for this project. There are no additional conflicts of interest to report.
Footnotes
ACKNOWLEDGMENTS
We thank Dr. Randy D. Blakely and Meagan Quinlan in Florida Atlantic University for recommending the serotonin transporter antibodies. Human brain tissues were obtained from the New York Brain Bank (NYBB) at Columbia University/Taub Institute, supported by NIH P50AG008702. YH effort was supported by Xiangya Third Hospital, Central South University.