
Abstract
Background:
The diagnostic workup for choreiform movement disorders including Huntington’s disease (HD) and those mimicking HD like phenotype is complex.
Objective:
The aim of the present study was to genetically define HD and HD-like presentations in an Indian cohort. We also describe HTT-CAG expansion manifesting as neuroferritinopathy-like disorder in four families from Punjab in India.
Materials and methods:
159 patients clinically diagnosed as HD and HD-like presentations from various tertiary neurology clinics were referred to our centre (CSIR-IGIB) for genetic investigations. As a first tier test, CAG-TNR for HTT was performed and subsequently HD-negative samples were screened for JPH3 (HDL2), TBP (SCA17), ATN1 (DRPLA), PPP2R2B (SCA12) and GGGGCC expansion in C9orf72 gene. Four families presenting as neuroferritinopathy-like disorder were also investigated for HTT-CAG expansion.
Results:
94 of 159 (59%) patients were found to have expanded HTT-CAG repeats. Pathogenic repeat expansion in JPH3, TBP, ATN1 and C9orf72 were not found in HD negative cases. Two patients were positive for SCA12-CAG expansion in pathogenic length, whereas 5 cases harboured TBP-CAG repeats falling in reduced penetrance range of 41– 48 repeats for SCA17. Four unrelated families, presented with atypical chorea and brain MRI findings suggestive of basal ganglia abnormalities mimicking neuroferritinopathy were found to harbour HTT-CAG expansion.
Conclusion:
We present SCA12 as a new reported phenocopy of HD which should be considered for diagnostic workout along with SCA17 for HD-like syndromes. This study also illustrates the necessity, to consider evolving HD like phenotype, as a clinical diagnosis for cases with initial manifestations depicting neuroferritinopathy.
Introduction
Huntington’s disease (HD) is an autosomal dominant, slowly progressive neurodegenerative disorder caused due to CAG trinucleotide repeat expansion on 5’end of exon 1 in HTT gene on chromosome 4p16.3 [1, 2]. The normal range of uninterrupted CAG repeat is 6– 26, intermediate alleles contain 27– 35 CAG repeats, whereas 36– 39 repeats shows reduced penetrance and more than 39 repeats are reported as pathogenic [3]. Increased number of CAG repeats in the HTT gene results in huntingtin protein with longer tract of polyglutamine amino acids causing abnormal interaction with other brain proteins leading to death of nerve cells [4]. The clinical manifestation of HD includes a distinct phenotype characterized by movement disorders mainly chorea, incoordination, cognitive deterioration and behavioral changes [5]. Prevalence of Huntington disease is high and estimated to be 3 to 7 per 100,000 in populations of Europe, North America and Australia whereas it appears to be less in other populations including Asia and Africa [6–8]. Nevertheless, the disorder has been reported to be frequent in India and parts of central Asia [9]. An increasing number of reports have shown that several neurodegenerative disorders may mimic the clinical presentation of HD. Approximately 1% – 7% of cases with symptoms and signs suggestive of HD do not show expansion mutation in HTT gene [10]. Different HD-like disorders namely HDL1, HDL2 and SCA17 have been recognized with different underlying genetic causes [11]. HDL2, caused due to expansion mutation in JPH3 gene (chromosome 16q24.2) is one such disease with indistinguishable HD phenotype but exclusively reported in patients with African ancestry. Normal range of CAG/CTG repeats for JPH3 is 6 to 28 whereas pathogenic, fully penetrant number of repeats reported so far is 41– 59 [12, 13]. Clinical overlap has also been demonstrated between HD and Dentatorubral-pallidoluysian atrophy (DRPLA) [14] and several forms of autosomal dominant ataxias [15] and therefore has been suggested to be considered for differential diagnosis for HD-like syndromes [16]. SCA12, although rare globally, is a very unique ataxia subtype prevailing in the Indian population [17]. Its clinical features include tremor and ataxia, however, its association with chorea phenotype remains to be explored. Neuroferritinopathy (NF) is another autosomal dominant basal ganglia disease with clinical features similar to those of HD [18]. Identification of the genetic basis of HD phenocopies is clinically important in the differential diagnosis of such cases.
This genetic heterogeneity prompted us to screen our cohort of patients for HD and associated disease phenotype. Since we conducted a genetic study solely, a thorough clinical data was either sparse or even missing in most cases with no family history available for many cases.
HTT-CAG screening was done for all cases included in the study. Subsequently, HD-negative samples were subjected to screening for TNR-CTG/CAG of JPH3 (HDL2), TNR-CAG/CAA of TBP (SCA17), TNR-CAG of ATN1 (DRPLA), TNR-CAG of PPP2R2B (SCA12) and GGGGCC repeats in C9orf72 genes. The repeat distribution of all alleles was also determined in the patient cohort. Clinical analysis was also performed for NF-like cases which harbored HTT-CAG mutation.
Methods
Subjects and participants
159 unrelated cases referred (from major Indian neurological clinics/academic institutes) with clinical diagnosis HD and its differentials enrolled in the CSIR-GOMED project was taken as the minimal inclusion criteria. All the included patients had chorea as major presenting feature. The study was approved by Institutes Human Ethic Committee. Written informed consent was obtained from all patients for molecular genetic testing and research. Peripheral venous blood samples were collected for DNA extraction using salting out method [19]. Our patient pool also included four kindred who were referred to us with the initial clinical diagnosis of NF with multiple symptomatic individuals (both males and females) with clinical features.
Limitation of the study design
As detailed clinical charting of features was not a part of the study, hence for the majority of the patients the diagnostic reliance was the major determinant of their inclusion for genetic investigations. Thus, thorough clinical data was sparse or even missing in most cases with no family history available. A further follow-up work of the study findings may be required for more detailed insight into each aspect categorically.
Genetic analysis
Estimation of repeat length distribution in HTT (HD), JPH3-CAG/CTG, TBP (SCA17), ATN1 (DRPLA) and PPP2R2B (SCA12) by flanking PCR
Fluorescent based PCR amplifying the triplet repeat region of HD, SCA17, SCA12, HDL2 and DRPLA was performed using appropriate primer pairs (Supplementary Table 1). PCR was done in a 10μl reaction containing 50 ng of genomic DNA, 0.4μM of each primer, 2.5 mM of each dNTPs, 0.4μM MgCl2 and 1 U of Taq Polymerase in the 5X buffer (Promega). PCR additives 0.25 M Betaine and 0.2 ul Formamide were used in the reaction for SCA12 and HD respectively. The fragment sizing and analysis of amplified and labeled PCR products were done on 3730XL genetic analyzer and GeneMapper software (Applied Biosystems, CA).
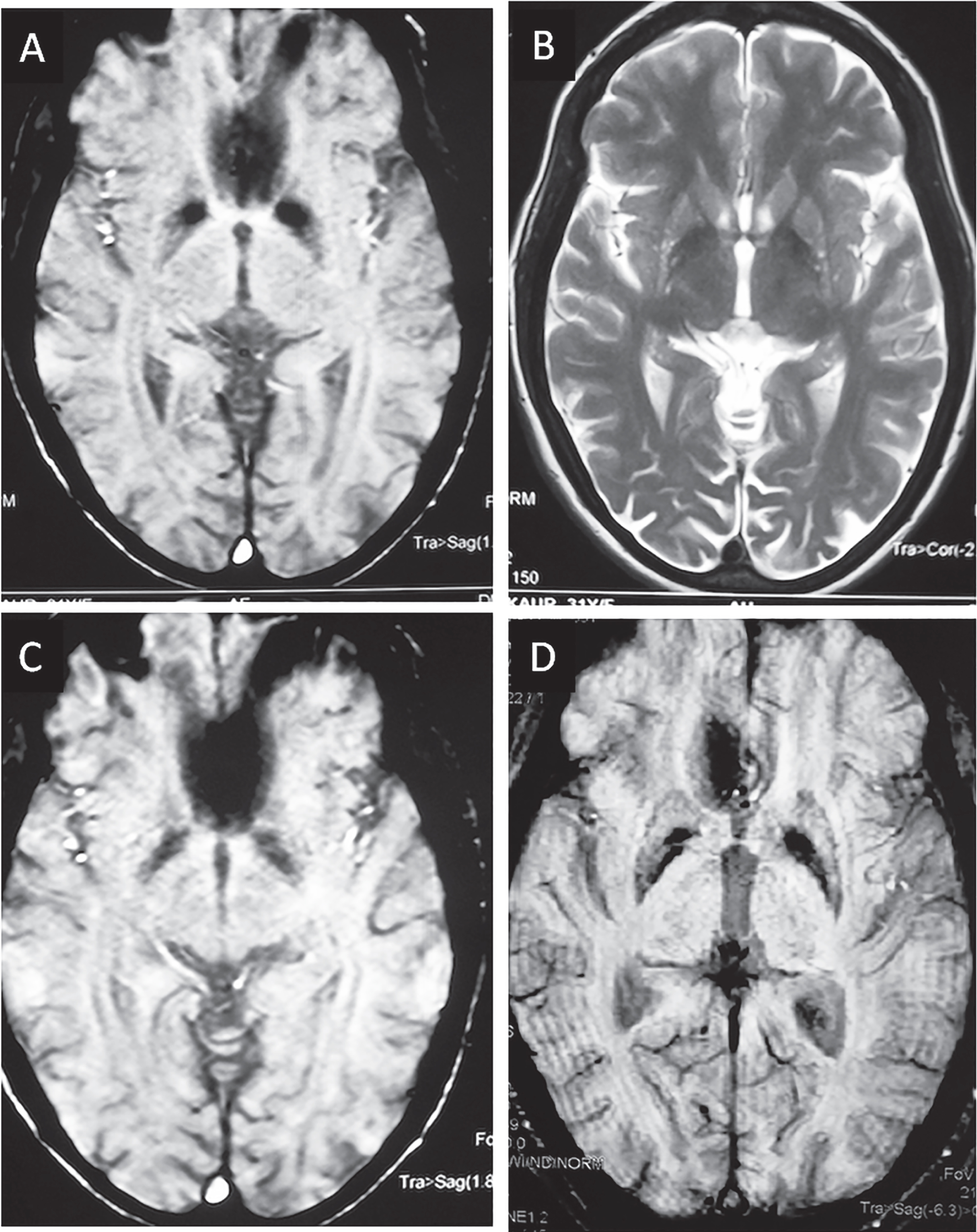
Brain imaging of HTT-CAG positive patients initially clinically diagnosed for suspected neuroferritinopathy. Family-1 (index patient) (A) Brain MRI, Axial Susceptibility weighted image showing hypointense signalling bilateral globi pallidi, substantia nigra and dentate nuclei. (B) T2 weighted brain axial image showing hyperintense signals in globus pallidi; Family-2 (index patient) (C & D) symmetrical hypointensity areas in bilateral globus pallidus on susceptibility weighted axial images.
Demographic details of N = 159 cases referred with clinical diagnosis of Huntington and its differentials. Tandem nucleotide repeat numbers obtained for HD and other related loci for all cases
RP-PCR for estimation of and G4C2 hexanucleotide repeat expansion in C9orf72 gene
A modified RP-PCR protocol was followed for the detection of the expanded allele of C9orf72. PCR was carried out in a 12 ml reaction, containing 100 ng DNA, 5 ml FailSafe G reagent (Lucigen), 2U G2 Hot start Polymerase (Promega), 0.25 mM 7-deaza-dGTP, 0.4 mM FAM-labelled forward primer, 0.125 mM reverse primer and 0.4 mM anchor tail reverse primer. Heat pulse extension PCR cycle was used for the amplification of C9orf72 repeat region. The fragment sizing and analysis of amplified and labeled PCR products were done on 3730XL genetic analyzer and GeneMapper software (Applied Biosystems, CA).
Analysis of FTL gene by Sanger sequencing
Mutation screening of FTL gene was performed in the probands of multiplex families with phenotype resembling NF. The entire coding region and intron-exon junctions of FTL gene was amplified by polymerase chain reaction with appropriate primer pairs. The amplified PCR products were subjected to Sanger sequencing using ABI 3730- genetic analyzer (ABI, Foster City).
Results
Brief clinical details of the study cohort
The patients included in the study were clinically diagnosed with HD and differentials with HD-like phenotypes. All n = 159 patients had chorea as major presenting feature. Of the total cases, n = 136 had generalized chorea/ choreoathetosis, n = 15 had chorea in addition to ataxia and n = 8 had chorea along with neurometabolic/NBIA like features. Of the total samples taken, 103 patients were males and 56 were females. Age of the patient cohort was in the range of 6– 74 years.
Clinical evaluation of affected individuals with chorea along with NBIA like features from four multiplex families was unique in neurological manifestations with variable age at onset (27– 34 years) of chorea and variety of extrapyramidal features, i.e., dystonia, dyskinesia (orolingual and oromandibular) and parkinsonian features. Few individuals had extraocular manifestations, vertical saccades and hypometric saccades. Behavioural changes were consistent in all the affected individuals. Multiple affected members from each of the 4 families was suggestive of dominant pattern of inheritance. Brain MRI (in selective subjects, Fig. 1) has showed changes suggestive of iron deposition in bilateral globus pallidum, substantia nigra and dentate nuclei on susceptibility weighted images (Fig. 1A– D).
HTT-CAG repeat analysis in the study cohort
Of the total 159 samples screened for HTT-CAG repeats, 59% (94/159) of cases showed expansion in the penetrance range (≥39 repeats) ranging from 39– 83 in our cohort. The youngest age of diagnosis was 15 years with 83 HTT-CAG repeats and oldest age was 74 years with 40 HTT-CAG repeats. Invariably, the maximum number of cases, 82% (77/94) had their CAG repeats in the range of 40– 50.
Categorically, 64% (87/136) of HD cases, 26% (4/15) of chorea with ataxia (as a differentials of HD) cases, 37% (3/8) of chorea with NBIA/neurometabolic features tested positive for HTT-CAG expansion (Table 1).
Analysis of tandem nucleotide repeats in other HD related loci (HDL2, SCA17, DRPLA, C9orf72 and SCA12)
Screening of all HTT-CAG negative cases for JPH3-CAG/CTG and ATN1-CAG did not show any large expansions falling in the pathogenic range. Similarly, no pathogenic expansion was observed in C9orf72. Two cases showed positive for large repeat expansion in PPP2R2B/SCA12 with the expanded allele carrying 56 and 57 CAG repeats. Since both the cases were clinically described as not only having chorea, but also ataxia, they were identified as SCA12 (Table 1). SCA17/TBP-CAG>41 was present in 4 cases of HD and 1 case of HD/ataxia group. The normal repeat range for JPH3 allele was 13– 24 showing bimodal distribution with maximum cases carrying CTG-20 repeats followed by CTG-23 repeats.
Screening for FTL mutations
A preliminary investigation of NF-like cases for non-synonymous mutations in FTL gene was essentially negative implying the absence of FTL mutations as genetic cause for the clinical symptoms observed in cases.
Discussion
An increasing number of choreiform movement disorders mimicking HD like phenotype has been identified to carry genetic defects other than HTT-CAG expansion. The HD-like disorders are majorly diseases, manifesting a predominantly choreatic syndrome and onset in adulthood or diseases presenting with predominant dystonia and Parkinsonism with onset in the first 2 decades [21]. Other genetic chorea forms have also been documented including chorea-acanthocytosis, NF, McLeod syndrome, benign hereditary chorea, Leysch Nyhan disease and Wilson’s disease [22]. Some non-genetic factors can also contribute to choretic movements as observed in vascular, rare autoimmune or infectious disorders as well as can be drug-induced [22]. Thus, it may be stated that these non-genetic factors might be responsible for the choretic movements in the HD negative cases from our patient cohort with missing family history.
We attempted to define the genetic role of different TNR expansions in causing HD clinical phenotype and related differential diagnosis, where chorea was considered as determining phenotype. We observed that HTT-CAG expansion mutation is the major determinant of progressive choreoathetoid phenotype in Indian patients. More than 50% of the cases showed expansion mutation in HTT gene. The screening of other TNR loci for all HTT-CAG negative HD and associated cases revealed no positive results except for PPP2R2B-CAG for SCA12 deliberating the near absence of other genetic causes in the Indian cohort.
Two most common HD-like syndrome associated with cerebellar involvement globally is SCA17 and DRPLA. A previous study by Wild et al., had shown SCA17 as the commonest neurogenetic diagnosis for HD phenocopies, [10] yet, we did not detect any cases for SCA17 with fully penetrant pathogenic repeat length for TBP-CAG. Five cases carried repeats which fell in the reduced penetrance range of 41– 48 for TBP-CAG. SCA17 repeat length in reduced penetrance range might account for the clinical features in the present cases although there are conflicting reports regarding the pathogenicity of TBP-CAG repeats in the range 41– 44 [23]. Furthermore, we did get two positive cases for another unique SCA subtype, SCA12, which is prevalent in India. The complete clinical details for these two patients were unavailable but they had been clinically diagnosed with chorea associated with cerebellar ataxia.
Various studies have been carried out in different countries for multiple genetic testing in HD phenocopy syndromes, revealing positive results from nil to 1– 2% [10, 11]. Predominantly, SCA17, JPH3 (HDL2) and C9orf72 positive cases has been identified in different populations. All patients reported till date for JPH3 (HDL2) have some African ancestry whereas C9orf72 expansion is the most common identified genetic cause of HD-like presentations in UK [13, 24]. It is imperative to state that these genetic loci were found negative in our patients and hence essentially missing from our cohort.
One of the crucial findings of this study was the presence of of HTT-CAG mutation in cases which were initially clinically diagnosed as NBIA-like disorder with symptoms of chorea and a neurometabolic case with dystonia.
NF, an autosomal dominant form of NBIA, characterized by slowly progressive, adult onset movement disorder presenting with chorea (50%) or dystonia (43%) or parkinsonism (7.5%) and is often associated with heterogeneous cognitive and neuropsychiatric phenotype [25, 26]. The early neuropathological features show iron deposition in the brain, and the basal ganglia are the most markedly affected. NF is considered a phenocopy of HD since multiple clinical features distinctive for NF have been found in HD cases too. We hereby report the presence of HTT-CAG mutations in multiple affected members from four families coming from similar geographical background in India, which were initially clinically diagnosed as NBIA like disorder. The clinical phenotype of the index cases was akin to NF, with hyperkinetic-asymmetrical and generalized choreic movements along with mild dystonia. Oral dyskinesia, dysarthrophonia and bradykinesia were commonly found among affected cases, further supporting NF like symptoms [27, 29] and hence initial diagnosis. Neuro-imaging features suggested iron deposition in bilateral globi pallidi, substantia nigra and dentate nuclei, another hallmark feature of NF [27]. Nevertheless, studies have also shown iron deposition in the pallidum, putamen and caudate in pre-HD and symptomatic-HD groups [30], indicative of HD like phenotype for such cases.
Alterations in brain iron homeostasis leading to iron accumulation has contributed notably in different neurodegenerative diseases. In the present context, when considering NF and HD, both seems to manifest deposition of iron in different regions of the brain during the course of pathogenesis. In NF, iron deposition in the form of dense ferritin-Fe spheroid inclusions are distinctly observed in different brain regions including dentatenuclei, globus pallidus, putamen, caudate, thalamus, and red nuclei [31]. Whereas, in HD, alterations in brain iron metabolism have been demonstrated within specific brain regions undergoing neurodegeneration, with increased iron deposition in caudate, putamen, and cortex [31, 32]. Consequently, initial diagnosis based on clinical manifestations and MRI can be dilemmatic in clinical settings.
Henceforth, these case studies provide critical insights for considering either NF like symptoms or evolving HD like phenotypes, while making clinical diagnosis for similar case presentations. Genetic testing for HTT-CAG mutations should be deliberated in such cases.
Conclusion
The present study describes SCA12 as a new HD phenocopy for the first time and also reports 5 HD phenocopies to be SCA17 mutation carriers in the incomplete or reduced penetrance range, whereas no C9orf72 cases have been detected. Of course, HD phenocopies are rare in India, but it is useful to investigate them if family history is positive or clinically suggestive. This study also brings forth the necessity to consider evolving HD like phenotype as a clinical diagnosis for cases with initial manifestations depicting NBIA.
Conflict of interest
The authors have no conflict of interest to report.