
Abstract
Background:
The relative contribution of grey matter (GM) and white matter (WM) degeneration to the progressive brain atrophy in Huntington’s disease (HD) has been well studied. The pathology of the spinal cord in HD is comparatively less well documented.
Objective:
We aim to characterize spinal cord WM abnormalities in a mouse model of HD and evaluate whether selective removal of mutant huntingtin (mHTT) from oligodendroglia rescues these deficits.
Methods:
Histological assessments were used to determine the area of GM and WM in the spinal cord of 12-month-old BACHD mice, while electron microscopy was used to analyze myelin fibers in the cervical area of the spinal cord. To investigate the impact of inactivation of mHTT in oligodendroglia on these measures, we used the previously described BACHDxNG2Cre mouse line where mHTT is specifically reduced in oligodendrocyte progenitor cells.
Results:
We show that spinal GM and WM areas are significantly atrophied in HD mice compared to wild-type controls. We further demonstrate that specific reduction of mHTT in oligodendroglial cells rescues the atrophy of spinal cord WM, but not GM, observed in HD mice. Inactivation of mHTT in oligodendroglia had no effect on the density of oligodendroglial cells but enhanced the expression of myelin-related proteins in the spinal cord.
Conclusion:
Our findings demonstrate that the myelination abnormalities observed in brain WM structures in HD extend to the spinal cord and suggest that specific expression of mHTT in oligodendrocytes contributes to such abnormalities.
INTRODUCTION
The differential regional pattern of brain atrophy is a well-established neuropathological phenomenon in Huntington’s disease (HD) [1]. Comparatively, the pathological characteristics of the second major part of the central nervous system (CNS), namely the spinal cord, are less well documented in HD. Recent studies using magnetic resonance imaging (MRI) have revealed cervical spinal cord atrophy in premanifest and early-stage HD patients [2, 3], indicating that spinal degenerative processes occur years before the onset of clinical symptoms. Furthermore, spinal cord atrophy in HD patients was shown to correlate with motor symptoms, suggesting that spinal cord pathology could contribute to certain clinical symptoms [2]. In addition, evidence from animal models of HD has revealed mild degeneration of spinal cord axons in the R6/2 model [4], and an association between muscle atrophy and cervical spinal motoneuron loss in the BACHD model [5], further suggesting a potential involvement of spinal cord pathology in the disease.
Analyses of brain pathology have long established that both grey and white matter (GM and WM, respectively) brain structures are profoundly affected in HD. Abnormalities in WM microstructure and loss of WM volume have been documented in postmortem HD brains [6, 7]. MRI and diffusion tensor imaging have demonstrated alterations of WM regions in patients with HD and in premanifest gene carriers [8–10]. Similar findings were reported in animal models of HD, where abnormal connectivity between brain WM regions and thinner myelin sheaths were found well before neuronal atrophy and onset of neurological disabilities [11–14].
There have been relatively few assessments of spinal cord pathology. A recent MRI-based analysis of the corticospinal tract (CST), the major WM tract that carries movement-related information from the motor cortex to the spinal cord, found WM microstructural changes in the CST in pre-HD subjects which worsened in patients with manifest HD and correlated with motor symptoms [15]. Another study has revealed loss of myelinated fibers in cervical and, to a lesser degree, thoracic areas of postmortem HD spinal cord [16], suggesting that WM pathology in HD may extend to the spinal cord. However, the identification of the patient in this case study was based on symptoms and postmortem examinations, with a detailed analysis of the spinal WM in genetically-confirmed HD still lacking.
Here we sought to investigate spinal cord pathology in a mouse model of HD, and in particular WM and myelination abnormalities. We have previously shown that intrinsic mutant huntingtin (mHTT)-mediated deficits in oligodendroglia contribute to myelination abnormalities and behavioral manifestations in HD [11]. Oligodendrocytes are essential for maintaining axonal integrity and function, and their dysfunction can lead to axonal pathology and neurodegeneration [17]. In the present study, using histological and electron microscopy analysis of myelinated fibers, we specifically evaluated measures of WM and myelin structure as well as the impact of the inactivation of mHTT in oligodendroglia on these measures in the spinal cord of the full-length BACHD mouse model of HD.
MATERIALS AND METHODS
Animals
BACHD mice expressing the full-length human mutant HTT transgene containing two loxP sites flanking the exon 1 were used and maintained on the FVB/N strain. NG2-Cre mice were backcrossed onto the FVB/N background and then crossed to BACHD mice to generate BACHDxNG2Cre (BN) mice [11]. Male and female mice were housed with littermates of mixed genotype in groups of 2–5 on a 12 h light/ dark cycle with free access to food and water. All experiments were performed with the approval of the Institutional Animal Care and Use Committee (IACUC no.151067) at the Biomedical Sciences Institute (ASTAR) and in accordance with their approved guidelines.
Transmission electron microscopy
Mice (3 per genotype, all males) were transcardially perfused with 2.5%glutaraldehyde and 2.5%paraformaldehyde (PFA) in phosphate buffered saline (PBS) before post-fixing the brains overnight at 4°C in the same buffer. Cervical spinal cord segments (C1-C3) were microdissected and post-fixed in 1%osmium tetroxide/1.5%potassium ferrocyanide solution for 1 h at room temperature (RT). After washing with 1%uranyl acetate in maleate buffer, the samples were dehydrated, infiltrated with epon, embedded, and polymerized at 60°C for two days. Ultra-thin slices (100 nm) were cut before imaging on a transmission electron microscope. For image analyses, axon and myelin fibres diameters were measured using ImageJ. 12 images with a total of more than 300 axons were analyzed for each mouse. Myelinated axons were divided into two categories and classified as ‘regular shaped axons’ when they presented a circular, quasi-circular or elliptical shape, while remaining axons were classified as ‘irregularly shaped axons’ (dystrophic axons).
Immunohistochemistry and stereological measurements
Cervical spinal cord segments (C1-C3) from 12-month-old animals were microdissected and post-fixed overnight in 4%PFA before switching to 30%sucrose in 1× PBS. Spinal cord segments were sliced coronally into 25μm coronal sections on a cryostat (Leica CM3050S) and were collected free-floating in PBS with 0.01%sodium azide. A series of sections spaced 300μm apart were used for the analysis. Sections were incubated with 1%phenylhydrazine in 1 x phosphate buffered saline (PBS) for 45 min at RT, subsequently washed 3 times with PBS and blocked in 5%normal goat serum (NGS) in PBS containing 0.1%Triton X-100 (TX) for 30 min at RT. Sections were then incubated with primary antibodies in 1%NGS in PBS-TX overnight at 4°C. Rabbit anti-Olig2 at 1:750 (Millipore AB9610), rabbit anti-GSTpi at 1:3000 (MBL, 311), rabbit anti-NG2 at 1:500 (Sigma-Aldrich, AB5320), and rabbit anti-Sox10 at 1:500 (Millipore, AB5727) were used. Sections were washed 3 times with PBS followed by incubation with biotinylated anti-rabbit antibody (1:200; Vectastain ABC HRP Kit, PK-4006; Vector Laboratories, Burlingame, CA, USA) for 1.5 h at room temperature with 1%NGS, 0.2%Triton X-100 in PBS. After three washes in PBS, sections were incubated in Vectastain Elite ABC reagent (Vector Labs Inc., Burlingame, CA, USA) for 2 h at RT and staining was visualized using 3,3′-diamino- benzidine (DAB) (ImmPACT DAB Peroxidase Substrate, SK-4105; Vector Laboratories, Burlingame, CA, USA). Slices were mounted on slides and images of whole spinal cord cross-sections were captured using a Nikon Ni-E upright microscope. The GM and WM area of the spinal cord was measured by tracing the perimeter of the respective areas using ImageJ and the total number of cells was counted. For GM and WM area, Olig2 staining was used and half of the spinal cord was measured to estimate total area and total cell counts.
Spinal cord tissue collection for molecular analysis
Cervical spinal cord segments (C4-C6) from 12-month-old animals were microdissected, snap-frozen in liquid nitrogen and stored at –80°C until further use.
Protein analysis
Protein lysate of cervical spinal cord segment (C4-C6) from male and female mice were prepared using RIPA buffer (Sigma-Aldrich) with 1 mM PMSF (Sigma-Aldrich), 5μm Z-VAD (Promega), 1 mM NaVan (Sigma-Aldrich), and 1× Complete Protease Inhibitor Cocktail tablets (Roche). For immunoblotting 30μg of protein lysates were separated on 12%Bis-Tris protein gel (Novex) with 20X MES running buffer (Novex) and transferred on nitrocellulose membranes. Samples were successively blocked with Odyssey Blocking Buffer (LI-COR, P/N 927-40000) 1 h at RT, and then incubated with primary antibodies at 4°C overnight. The following primary antibodies were used: Ermin (1:1000; Merck), MBP (1:500; Millipore), MAG (1:2000; Millipore), Sept8 (1:500; Proteintech), β-actin (1:5000; Sigma-Aldrich) and Calnexin (1:5000; Sigma-Aldrich). Membranes were then incubated with secondary antibodies (1:10,000) 2 h at RT: Alexa-Fluor goat anti-rabbit 800, Alexa-Fluor goat anti-mouse 680, Alexa-Fluor goat anti-rabbit 680, and Alexa-Fluor goat anti-rat 800 (all from Life Technologies). The membrane was imaged using the LiCor Imaging System and Odyssey V3.0 software (LiCor), followed by intensity analysis with ImageJ.
RESULTS
White, but not grey, matter atrophy in the spinal cord is rescued by inactivation of mutant HTT in oligodendroglia
To determine whether GM and WM regions are atrophied in the spinal cord of 12-month-old BACHD mice, we measured the area of GM and WM in the cervical (C1-C6) regions of the spinal cord by histological assessment using the oligodendroglia marker Olig2 (Fig. 1A,C). The spinal total area, GM, and WM area were significantly lower in BACHD mice compared to WT controls (Fig. 1D-F), demonstrating atrophy of GM and WM in spinal cord of HD mice.
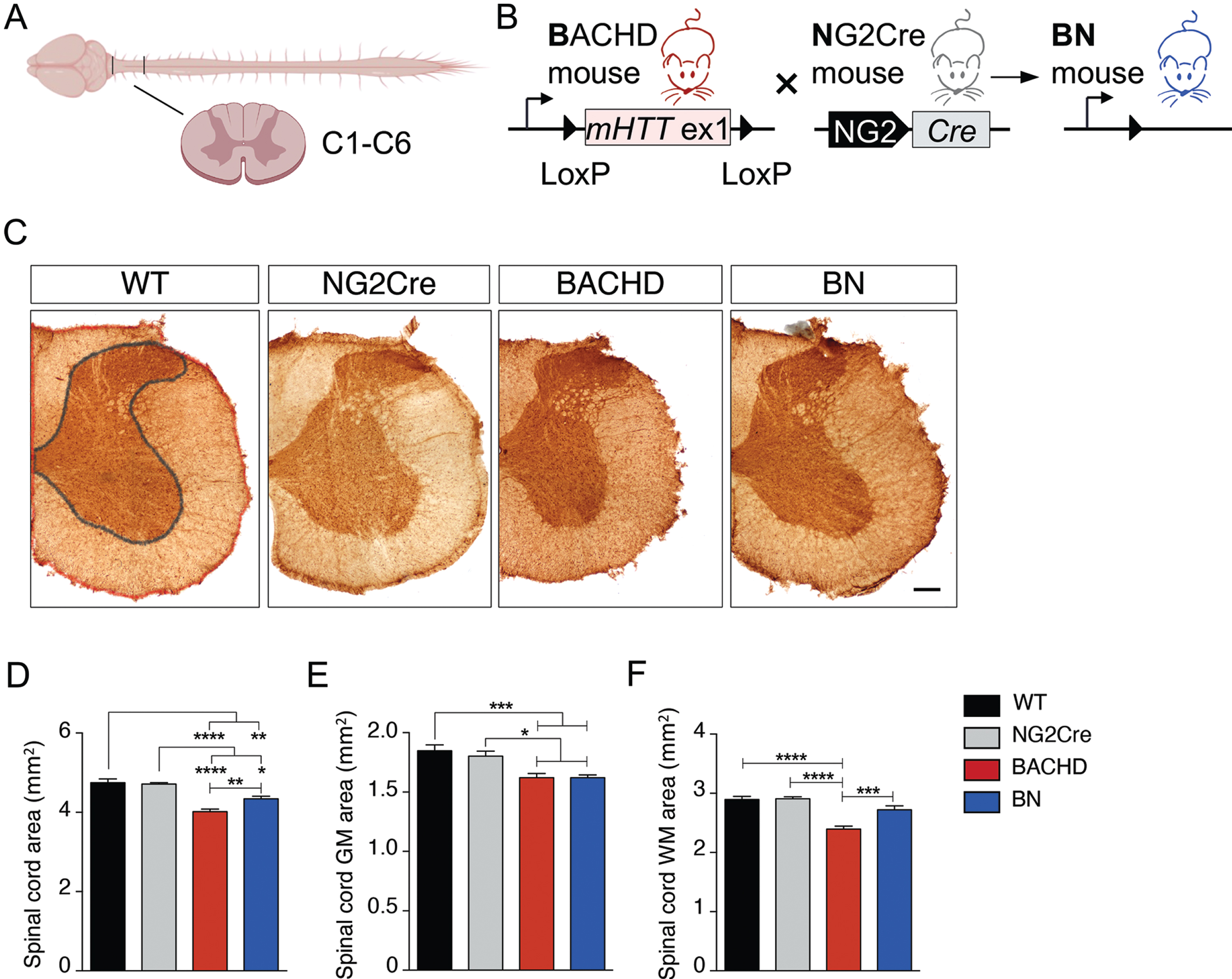
OPC-intrinsic effects of mHTT on white, but not grey, matter spinal cord in HD mice. A) Spinal cord diagram. B) Schematic representation of Cre-mediated genetic reduction of mHTT expression in OPCs (NG2 + cells) in BACHD mice. C-F) Spinal grey matter (GM), white matter (WM), and total area were evaluated measuring half side of the spinal cord using Olig2 staining in 12-month-old mice. C) Images of cervical spinal cord sections at 12 months of age. WT image shows how area was measured. GM area is depicted in grey, total area is depicted in red, and WM area was calculated subtracting GM area from total area. Scale bar represents 200μm. D-F) Spinal total area is decreased in BACHD and BN mice compare to WT (D), but it also shows a significant increase in BN mice compared to BACHD (D). BACHD mice present significant decrease in (E) GM and (F) WM compared with WT mice. Spinal WM, but not GM, area is rescued in BN mice (E,F). N = 6–13 per genotype. Data represent means±SEM; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by one-way ANOVA followed by Tukey’s test.
We have previously shown that callosal WM abnormalities in the BACHD are caused by intrinsic effects of mHTT in oligodendroglia [11]. To examine whether the spinal WM atrophy observed in BACHD is similarly caused by intrinsic mHTT-induced oligodendroglial defects, we used the previously described BACHDxNG2Cre (BN) mouse line where mHTT is specifically reduced in oligodendrocyte progenitor cells (OPC) (Fig. 1B) [11]. The total spinal area in BN mice was significantly lower than WT mice, but also significantly higher than BACHD mice, indicating partial rescue (Fig. 1D). To further explore the partial rescue in total spinal area, assessment of spinal GM and WM areas in BN mice was performed. We showed that white, but not grey, matter atrophy in the spinal cord of BACHD mice was rescued in BN mice (Fig. 1E,F). Consistent with this finding, we observed significant atrophy of the spinal GM region in BN mice compared to WT mice (Fig. 1E), but no differences in spinal WM area (Fig. 1F). Together these findings demonstrate that WM abnormalities in HD extend to the spinal cord and are primarily driven by cell-intrinsic defects of mHTT in oligodendroglia.
Oligodendroglia-intrinsic effects of mHTT cause myelin deficits in the spinal cord of BACHD mice
To test whether the myelin deficits observed in the corpus callosum also extended to the spinal cord, we used electron microscopy to visualize myelinated fibers in the cervical area of the spinal cord at 12 months of age (Fig. 2A) and analyzed the g-ratio. G-ratio of myelinated axons, used as a measure of myelin thickness, was calculated as the ratio of axon diameter to myelinated fiber diameter (Fig. 2B). The mean g-ratio of myelinated axons was significantly higher in BACHD mice compared to WT mice (Fig. 2C,E,F), indicating that their myelin sheaths were thinner. Consistent with the increased spinal WM area observed in BN mice, we found thicker myelin sheaths in BN mice as shown by a decrease in g-ratio. These results indicate that selective reduction of mHTT in oligodendroglia rescues the myelin thickness deficits not only in brain but also spinal cord WM (Fig. 2D,E,F). To exclude that the changes observed in the g-ratio are due to differences in axonal diameters, we analyzed axonal diameter frequencies and found that distribution of axonal diameters was similar between genotypes (Supplementary Figure 1A,B), indicating that differences in g-ratio between BACHD and BN mice are indicative of changes in myelin thickness and not the result of changes in axonal caliber. We also analyzed the number of dystrophic axons and did not find significant differences among the genotypes (Supplementary Figure 1C).
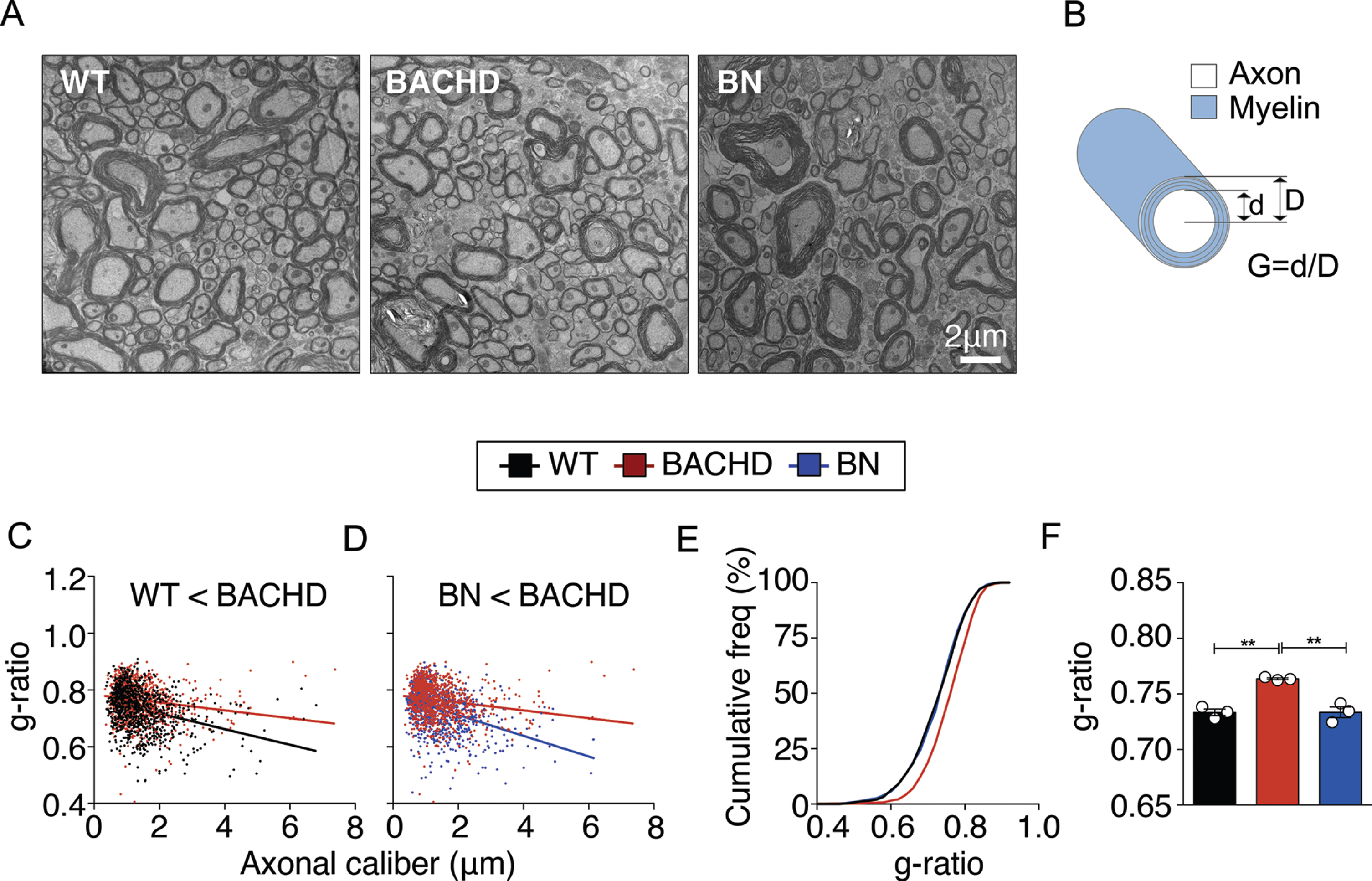
OPC-intrinsic effects of mHTT cause myelination abnormalities in the spinal cord of HD mice. A) EM images of myelinated axons in cervical spinal cord sections at 12 months of age. Scale bar represents 2μm. B) G-ratio calculation diagram. C-F) Higher g-ratios (thinner myelin sheaths) in BACHD mice are rescued in BN mice. N = 3 per genotype; ∼300 axons quantified per animal. Data represent means±SEM; **p < 0.01 by one-way ANOVA followed by Tukey’s test.
Inactivation of mutant HTT in oligodendroglia rescues deficits in myelin-related proteins in the spinal cord
In order to evaluate whether the rescue of WM atrophy and myelin thickness in the spinal cord seen with inactivation of mHTT in oligodendroglia in BN mice is associated with changes in myelin protein levels, we next examined the expression levels of the myelin-related proteins Ermin, myelin basic protein (MBP), myelin-associated glycoprotein (MAG), and Sept8 by immunoblotting. We found that Ermin, MBP, and MAG were significantly increased in BN versus BACHD mice (Fig. 3A-C), whereas Sept8 did not show any significant changes (Fig. 3D). This increase in the levels of myelin-related proteins is consistent with the rescue in myelin thickness observed in BN mice.

Myelin protein expression in BACHD is mediated by OPC-intrinsic effects of mHTT. A-D) BN mice present significant increase in (A) Ermin, (B) MBP, (C) MAG, but not (D) Septin-8 protein expression levels compared to BACHD mice in the spinal cord at 12 months of age. N = 10–11/genotype in A,B or N = 8–9 in C or N = 13 in D. All bands were used for quantification. Data represent means±SEM; *p < 0.05; two-tailed unpaired Student’s test in B (p = 0.0273, t = 2.391, d.f. = 19), or one-tailed unpaired Student’s test in A (p = 0.0354, t = 1.909, d.f. = 20), C (p = 0.0462, t = 1.798, d.f. = 15), and D.
No effect on oligodendroglial populations in BACHD or BN mice
To investigate whether the spinal WM atrophy and myelination abnormalities in BACHD mice and the rescue observed in BN mice are associated with changes in oligodendroglia populations in the spinal cord, we analyzed the density of relevant cellular populations by stereology assessment using different oligodendroglia lineage markers, namely Olig2 (a transcription factor expressed throughout the entire oligodendrocyte lineage), GST-pi (a mature myelinating oligodendrocyte marker), and NG2 (an OPC marker). Consistent with our previous observations in the brain, no changes in the density of these oligodendroglia populations were observed in the spinal cord of BACHD and BN mice at 12 months of age (Supplementary Figure 2).
DISCUSSION
In this study, we characterize for the first time the WM and myelination abnormalities in the spinal cord of HD mice and show that they are primarily driven by cell-intrinsic defects of mHTT in oligodendroglia. Specifically, we first show that BACHD mice present spinal GM and WM atrophy and thinner myelin sheaths compared to WT mice at 12 months of age, suggesting the involvement of spinal cord degeneration in the pathology of HD. Second, we show that specific inactivation of mHTT in oligodendroglia rescues the WM phenotypes, providing clear evidence that oligodendrocytes contribute to the spinal WM pathology of HD.
We have previously shown that callosal myelination abnormalities are an early and progressive feature of HD and are associated with impaired white matter plasticity in response to acute oligodendrocyte injury [18] and environmental interventions [19] in HD mice. We have further shown that inactivation of mHTT in oligodendroglia population alone is not sufficient to improve striatal neuropathology in HD mice. Here we show that spinal GM atrophy in BACHD mice is similarly not rescued after mHTT reduction in oligodendroglia. Spinal GM mainly consists of cell bodies of interneurons, motor and sensory neurons, that still express mHTT in BN mice, and therefore contribute to their degeneration. In addition, axons of spinal GM are largely unmyelinated, indicating that oligodendrocytes may not play a major role in their survival.
One of the major WM pathways that carries movement-related information from the primary motor cortex to the spinal cord is the corticospinal tract. Damage to the corticospinal tract has been associated with a reduced motor control, together with other symptoms known as upper motor neuron syndrome. Corticospinal tract connectivity is altered in HD patients and correlates with motor scores in HD patients [15]. As previously mentioned, spinal cord atrophy also exists in HD patients and correlates with motor symptoms. These studies suggest that damage of spinal cord pathways together with spinal cord atrophy could contribute to certain clinical symptoms of the disease. BACHD mice mimic certain neurological features of the human condition and we have previously observed an improvement in motor and psychiatric-like functions after specific mHTT reduction in oligodendroglia [11]. This improvement could therefore reflect not only the rescue of WM callosal abnormalities, but also the rescue of spinal WM pathology in BN mice.
A noninvasive assessment of spinal cord damage is through the measurements of evoked potentials (EPs) by electroencephalography or electromyography. EPs are electrical signals that can determine whether there is motor or somatosensory impairment. Somatosensory evoked potentials (SEPs) and motor evoked potentials (MEPs) of the cortico-spinal system were found to be altered in HD patients [20–22], where MEP and SEP abnormalities correlated with motor symptoms and severity of illness respectively. Although it has been previously suggested that these alterations could be just a consequence of basal ganglia dysfunction, they may also reflect damage of the cortico-spinal tract, including spinal WM abnormalities. Indeed, EPs can be very helpful indicators of spinal cord demyelination [23], as well as a potential biomarker to track functional remyelination [24]. Although this suggests a possible correlation between altered EPs and spinal demyelination in HD, this relationship has not been evaluated to date. Future studies to investigate spinal WM pathology and any association with EPs in HD should be considered.
Beyond HD, some evidence of spinal pathology has been reported in other forms of neurodegeneration that are not generally associated with the spinal cord. For example, in both patients and transgenic mouse models of Parkinson’s disease, degeneration of spinal WM tracts as well as breakdown of myelin sheaths have been reported [25–27]. Similarly, in Alzheimer’s disease (AD), abnormal phosphorylation of tau protein and deposition of amyloid-β protein were found in the spinal cord of AD patients and animal models of AD [28]. Altogether, these reports provide new evidence that spinal cord pathology including WM degeneration may play a broad role in neurodegenerative disorders.
Collectively, our present study suggests that spinal cord degeneration is involved in the pathogenesis of HD. Further studies are needed to better elucidate the mechanisms underlying spinal degeneration and to understand whether interventions directly targeting spinal cord pathology specifically, and white matter degeneration more broadly, may be of therapeutic benefit in HD.
Footnotes
ACKNOWLEDGMENTS
C.F.B. is supported by a fellowship from the Hereditary Disease Foundation. The work was partly funded by grants from the Agency for Science Technology and Research (Singapore) and the National University of Singapore to M.A.P.
CONFLICT OF INTEREST
The authors have no conflict of interest.