
Abstract
Huntington’s disease (HD) is a devastating neurodegenerative disorder that urgently needs disease-modifying therapeutics. To this end, collaboration to standardize clinical research practices in the field and drive progress in addressing drug development challenges is paramount. At a meeting in 2017 organized by CHDI Foundation and the Critical Path Institute, stakeholders across the pharmaceutical industry, academia, regulatory agencies, and patient advocacy groups discussed the need for and potential impact of a consortium dedicated to HD regulatory science. Consequently, the Huntington’s Disease Regulatory Science Consortium (HD-RSC) was formed, a precompetitive consortium that is dedicated to building a regulatory strategy to expedite the approval of HD therapeutics.
Keywords
INTRODUCTION
Huntington’s disease (HD) is an autosomal-dominant neurodegenerative disorder caused by expansion of a CAG repeat in the huntingtin gene that encodes a mutant huntingtin (mHTT) protein and is characterized by devastating motor impairment, cognitive decline, and neuropsychiatric disturbances [1]. Recent scientific advancements that improve further understanding of HD pathophysiology provide an unprecedented opportunity to develop therapeutic interventions to markedly slow disease progression [2, 3]. Consequently, there is now a robust HD therapeutic landscape with many approaches targeting relevant pathologic pathways, including lowering the amount of mutant huntingtin. While approval of successful treatments remains elusive, data from studies conducted to date still promise to be scientifically revealing with the potential to facilitate drug discovery and development. However, drug developers still face many challenges in advancing HD therapeutics; importantly, this includes coming to agreement on and then meeting appropriate regulatory requirements.
In November 2017, the Critical Path Institute (C-Path; a nonprofit organization that establishes public-private partnerships between various stakeholders to develop new methods to assess the safety and efficacy of medical products) and CHDI Foundation (CHDI; a privately-funded nonprofit biomedical research organization exclusively dedicated to collaboratively developing therapeutics that substantially benefit those affected by HD) co-organized a meeting of representatives from the pharmaceutical industry, academia, regulatory agencies (the FDA and the EMA), and patient advocacy groups to discuss the regulatory science advancements required to build an expedient path to HD therapeutic approval.
A consortium research plan was presented at this launch meeting that outlined the proposed scope of activities and working group goals of the HD-RSC, including data standards development, unified database creation, disease progression modeling, evaluation of biomarkers (particularly biofluid and imaging biomarkers) and clinical outcome assessments, and the potential for preparation for regulatory acceptance of these measures. The HD-RSC goals of advancing biomarkers, disease modeling, and outcome assessments represent tangible tools to efficiently advance new drug candidates. Regulatory agency representatives acknowledged the unparalleled opportunity that HD represents within neuroscience due to its monogenic nature and agreed that the HD-RSC represents a framework to enable alignment and collaboration.
The HD-RSC was officially launched in March 2018 under formal membership agreement, and by 2021 has grown to consist of 37 members from biotechnology, pharmaceutical, technology, nonprofit biomedical research organizations, and patient advocacy groups, along with participation from academic individuals (see Fig. 1 for membership as of December 2021 and “Future Directions” section below for information regarding membership beginning in 2022). Here we outline the organization of the HD-RSC and report on the activities that have occurred since inception and the strategy for its ongoing work.
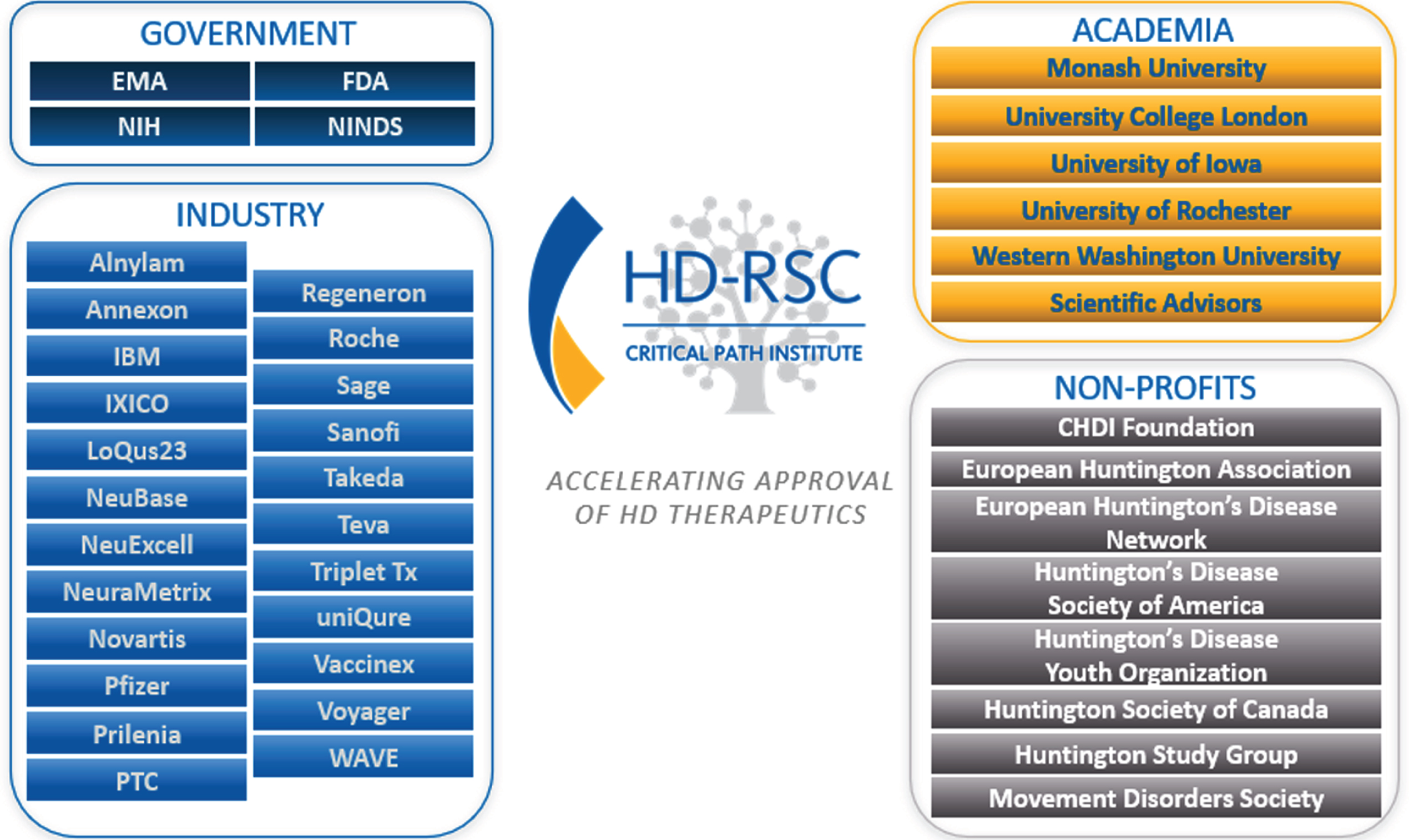
HD-RSC contributors as of December 2021 (includes industry, academic, and non-profit members).
CONSORTIUM GOVERNANCE STRUCTURE
The HD-RSC governance includes representatives from a diverse range of stakeholder organizations. From its initiation until mid-2021, the HD-RSC was funded by CHDI Foundation. During that time, leadership of the HD-RSC comprised of co-directors from C-Path (2), CHDI (2), academia (1), and industry (1), who provided strategic oversight for consortium activities. Additionally, a Coordinating Committee consisting of 1–2 representatives from each of the member organizations (plus scientific advisors who participate independent of their respective organizations) convenes monthly to assess working group progress and deliberate on the consortium portfolio.
In the HD-RSC’s first three years, work to accomplish the preliminary consortium objectives was driven by specific working groups (see below). Members sent representatives from their respective organizations to attend and participate in working group meetings, with each member organization represented in at least one working group.
ALIGNMENT WITH REGULATORY AGENCIES
The engagement of the FDA [4] and EMA (and other regulatory agencies as appropriate) is crucial to the success of all C-Path activities. The goals and objectives of the HD-RSC are aligned with consortium member and stakeholder priorities and needs, including the regulatory agencies and capitalize on C-Path’s core competencies and regulatory success in other disease areas; this includes the FDA designation of a trial simulation tool for mild to moderate dementia of the Alzheimer’s disease (AD) type as fit-for-purpose [5] as well as the EMA qualifications of DAT imaging as an enrichment biomarker for Parkinson’s disease [6] clinical trials and the development of a clinical trial simulator in pre-dementia [7]. During the early formation of the HD-RSC, the regulatory representatives emphasized that the HD-RSC should focus on disease prevention since intervening early in the underlying pathology will have the greatest benefit for HD families. Consequently, incorporating biomarkers and outcome measures as key covariates into models and tools are important consortium goals.
Patient-focused drug development is critical for HD. This requires both patient and caregiver input to evaluate therapeutics and the patient experience, since cognitive impairment often alters patient insight. The FDA’s patient-focused drug development meeting on HD in 2015 and Voice of the Patient report are tangible examples of this kind of outreach to the patient community [8]. Crucially, patient advocacy organizations are also members and key constituents of the HD-RSC, and these representatives regularly participate in the monthly Coordinating Committee meetings to ensure that HD-RSC activities are aligned with patient needs. The HD-RSC also interacts with The Huntington’s Disease Coalition for Patient Engagement (HD-COPE), an international collaboration between patient advocacy groups formed to ensure the patient voice is heard in HD therapeutic development.
DATA WORKING GROUP
Development of CDISC standards
Quality clinical trial and observational study data serves as the cornerstone of all tools that are developed by C-Path. The ability to readily understand and use such data requires the development of controlled terminology and defined standards that optimize data capture and structure. CDISC standards help enable data usability and quality control by systematizing data elements that are common across the majority of clinical research studies, and these standards are now required by the FDA for all new drug submissions to help efficient processing, review, and archiving of submissions. Furthermore, CDISC standards are especially valuable for rare diseases where studies/trials are often limited in size; they facilitate data harmonization and integration into larger datasets to provide a quantitative understanding of disease natural history, which will ease common trial design challenges and enable a path to faster regulatory approvals [9].
Accordingly, HD CDISC standards and the companion Therapeutic Area User Guide for HD (HD-TAUG) were developed in a collaboration between CDISC and the HD-RSC and published in October 2018 after review periods for public comment. HD key opinion leaders from academia and industry were consulted during this development process to determine the common data elements, variables, and concepts to be included in the HD-TAUG. The measures selected for CDISC standards development included genetics as well as imaging and biofluid biomarkers.
Integrated HD clinical database
To accelerate HD therapeutic development, it is essential for the community to have access to patient-level data generated through both observational studies and interventional clinical trials [10], so a key objective of the Data Working Group is to acquire, standardize, and aggregate clinical datasets to create a comprehensive HD Database within the C-Path Online Data Repository (CODR) using the CDISC HD-TAUG standards (Fig. 2). The HD-RSC identified the most important datasets at launch (Fig. 3), and there is commitment from consortium stakeholders to make them available.
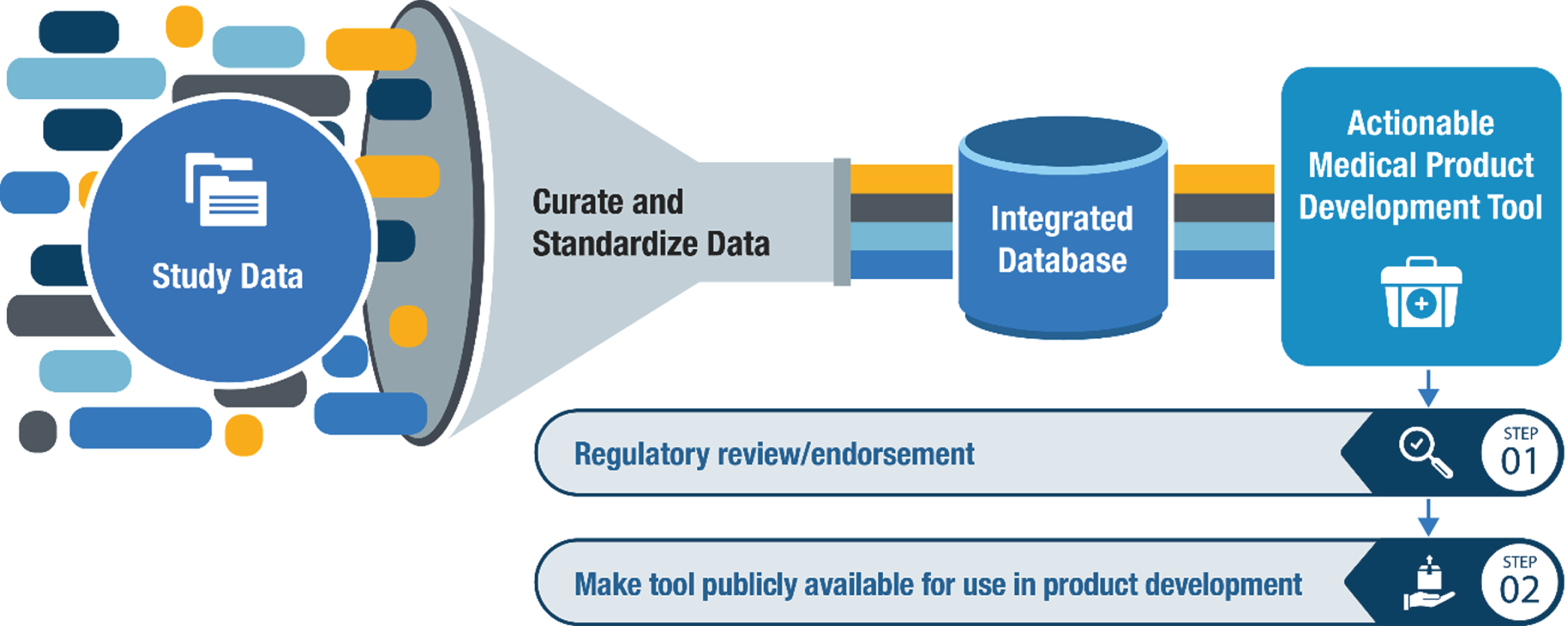
Pathway for developing novel Medical Product Development Tools for a proposed Context-of-Use (COU).
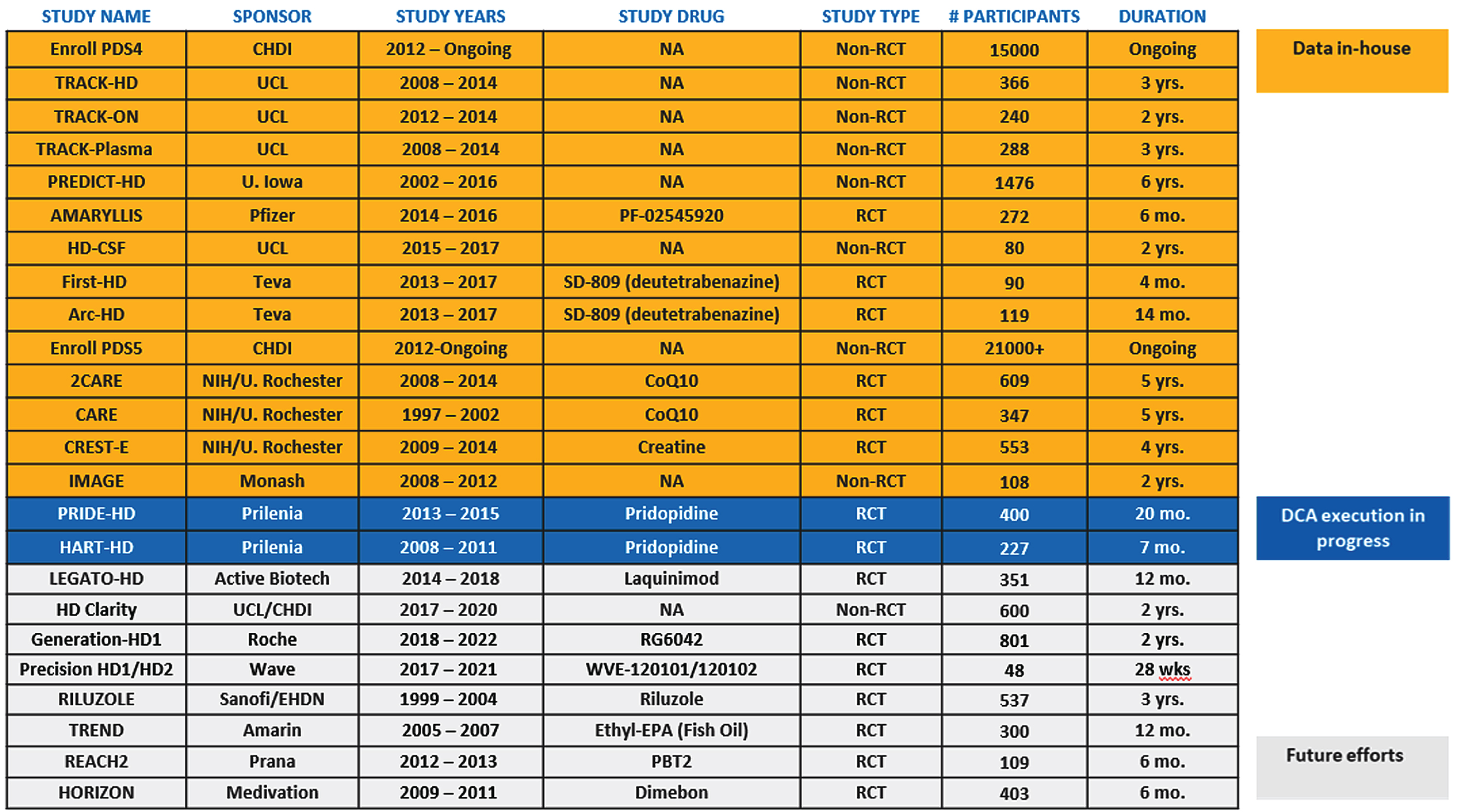
HD-RSC prioritized data inventory.
This comprehensive HD Database will include data related to clinical outcome measures in all relevant disease domains (including motor, cognitive, and behavioral), biological measures of disease (such as brain region volumes and HTT quantification in CSF), and participant characteristics (such as genetic information regarding CAG-expansion length and other disease modifiers). The Modeling and Simulation Working Group is using this data to build drug-development tools, such as quantitative disease progression models and clinical trial simulation (CTS) platforms (see below) that will provide insight into the longitudinal dynamics of biomarkers and clinical outcome assessments over the disease course and help integrate these measures into clinical trials.
In addition to supporting consortium research objectives, this HD Database will also be accessible to the community in the form of an integrated database, with the level of access determined by each data contributor. Important HD datasets have already been acquired by the consortium, including from industry-sponsored clinical trials (Fig. 3). Work to facilitate access to the HD Database for qualified researchers is ongoing and will be made available in 2022.
WORKING GROUP PRIORITIZATION
Modeling and simulation
The Modeling and Simulation Working Group was initiated with two primary objectives: 1) to develop a disease progression model capturing longitudinal change of clinical outcome measures and biomarkers throughout the disease process, and 2) to build a quantitative CTS platform based on the disease progression model [11]. The Working Group will take a stepwise approach, beginning with the disease period following clinical motor diagnosis since datasets from this phase are the most readily available.
Similar to the precedent in AD [5, 11], the Modeling and Simulation Working Group has developed a comprehensive disease-progression model and is currently developing a CTS tool to optimize clinical trial enrichment and the design of efficacy studies in HD. As data allows, the CTS tool will include a placebo-effect model, drug-effect model, and a participant drop-out model to optimize study design, management, and decision making. These models will be designed to provide critical information to industry stakeholders regarding enrichment strategies, cohort size, and trial duration required to meet a specified outcome. The CTS tool will be continually refined and updated as more data becomes available to the HD-RSC, especially to include participants who are earlier in their disease course.
The Modeling and Simulation Working Group has initiated a formal submission for its clinical trial simulation tool to the FDA’s Fit-for-Purpose Initiative following the submission of a Letter of Intent to the Agency in December 2020, which included the proposed context-of-use statement, an overview of the data sources, the proposed modeling analysis plan, and questions for the agency. In response to the Letter of Intent, the HD-RSC held a pre-submission call with the FDA in February 2021 and received formal feedback on this submission. At the time of publication of this manuscript, a briefing document has been submitted to the Office of Clinical Pharmacology at the FDA (July 2021), triggering the next phase of the official regulatory review.
Biomarkers
A biomarker that has clear clinical significance could greatly advance HD therapeutic development by catalyzing progress and de-risking programs. Biofluid and imaging biomarkers are in development and under investigation, but more work is required (even though vMRI has amassed substantive supporting evidence [12, 13]) to demonstrate their regulatory readiness and establish how they relate to clinical endpoints over time and in response to intervention. Data from interventional clinical trials will be critical to establishing these relationships and integrating this work with the Modeling and Simulation Working Group efforts will be instrumental in ultimately supporting regulatory endorsement, acceptance, or qualification.
Evaluating an intervention’s ability to prevent or delay the first signs and symptoms of HD will require biomarkers that correlate with very early indications of clinical benefit and that can assess therapeutic efficacy. The Biomarker Working Group has surveyed the biomarkers currently used or in development for use in HD clinical trials and assessed their regulatory-ready status based on the current needs in HD drug development, the viability of leading candidates, and the access to relevant data. The group is initially focused on leading candidates— mutant or total HTT in CSF, and caudate volume— due to their significant interest to the field, level of study, and biologic plausibility. More specific regulatory goals for these candidates will be determined as additional data is generated and shared with the consortium.
The current focus of this group is to establish industry-wide consensus practices for acquisition and analysis of these biomarkers to facilitate regulatory review, interpretation of results, as well as enable integration of biomarker data into aggregated datasets for analysis, including in the Modeling and Simulation Working Group. To that end, a biofluids sub-group has discussed the successes and challenges of key assays used in HD clinical research and is comparing mutant and total huntingtin assays in use or in development to converge on recommendations for assay harmonization.
Imaging experts from this working group have published two papers that focus on the use of vMRI-based biomarkers in HD; an evidentiary review [12] and a companion publication regarding optimized use of vMRI in HD clinical trials [13]. The latter proposes four core recommendations to address vMRI standardization in HD research: a checklist of standardized practices for the use of vMRI; targeted projects to evaluate advanced vMRI methodologies; the definition of standard MRI-based anatomical boundaries for key brain structures; and broad access to raw images and derived data from HD clinical research. Future work in the Biomarker Working Group will focus on enacting these recommendations.
Clinical outcome assessments (COAs)
COAs that better measure HD domains are required to appropriately characterize and assess clinically meaningful change (i.e., a positive change in how a patient feels, functions, or survives) within all disease stages [14]. The Unified Huntington’s Disease Rating Scale (UHDRS), a clinician-rated COA that evaluates the motor, cognitive, behavioral, and functional capacity of individuals based on clinical presentation, is the current gold-standard in clinical trials. However, it is not sensitive in evaluating these domains in individuals very early or late in their disease course [15–17]. COAs that sensitively assess specific features in and across different stages of the disease are required. This would also enable future clinical trials to shift towards evaluating therapeutics earlier in the disease, perhaps prior to clinical motor diagnosis [18].
The COA Working Group has received iterative feedback from the FDA regarding two novel COAs under development, the Functional Rating Scale Taskforce 2.0 (FuRST 2.0) and the HD Cognitive Assessment Battery (HD-CAB) [19]. FuRST 2.0 is a function-based patient reported outcome assessment of daily-living experiences meant to evaluate the disease period surrounding HD clinical motor diagnosis. HD-CAB is a battery of six performance-based cognitive tests developed to evaluate cognitive changes in similar target populations. Validation of these COAs is underway.
A primary endpoint in a clinical trial evaluating individuals before or soon after clinical motor diagnosis would have to directly measure symptoms that are both meaningful to patients and sensitive to changes across affected domains. Therefore, the group is also evaluating novel measures of cognition that are performance based and may evaluate changes that are clinically meaningful to individuals with HD and their families [8, 20]. Additionally, the COA Working Group may explore the use of digital measures and technologies in HD, which are becoming more widely used in neurodegenerative indications [12–23] and may be more sensitive than current measures or fill an unmet measurement need for those affected by HD.
Regulatory science forum
Developing therapeutics that will prevent or delay the emergence of HD symptoms is an ultimate goal for the HD community. To conduct clinical trials in a population before clinical motor diagnosis, and eventually before the presentation of any signs and symptoms of HD, requires a detailed understanding of disease progression. This includes accounting for heterogeneity in HD presentation and understanding how to appropriately utilize clinical outcome assessments and biomarkers throughout the disease course.
The Regulatory Science Forum working group was formed in 2019 within the HD-RSC by inviting a representative from a pharmaceutical or biotech company, among the members of HD-RSC, to be a voting member if the company’s pipeline included a therapeutic candidate in active clinical development. Additionally, academic and physician scientists were selected to match the number of industry representatives and provide geographic diversity to capture patient considerations from the global HD community.
The Regulatory Science Forum has used formal consensus methodology to develop a new HD framework called the HD Integrated Staging System (HD-ISS) [24] that comprises a biological definition of HD based on the CAG-repeat expansion in HTT and an evidence-based staging system. This work is analogous to work in the AD field [25] that has catalyzed early-intervention trials in AD by focusing on biological factors rather than solely clinical signs and symptoms. Also similarly, the HD-ISS is primarily intended for research use and focuses on prognostic biological, clinical, and functional landmarks. Data-driven landmark thresholds define HD-ISS Stage boundaries using extreme values in models of the control population and are CAG independent. Observational datasets were used to internally validate the framework; using the HD-ISS in prospective research studies will now enable further validation.
A biological research definition of HD standardizes biological concepts and provides a common language to enable cohesive clinical research across the full course of the disease. Importantly, it is unconstrained by concepts such as “manifest,” “premanifest,” or “prodromal.” Ultimately, the HD-ISS framework will allow clinical trials evaluating therapeutics that target HD biology earlier in the disease, perhaps even prior to the onset of clinical symptoms or functional impairment. The Regulatory Science Forum received informal feedback during the consensus process from regulatory agencies and the patient community and will continue to align with these key partners in the future. After publishing the HD-ISS, the next step for the working group will be to analyze and nominate enrichment biomarkers and outcome assessments appropriate for each Stage and across Stages with the goal of advancing regulatory science in support of drug development across the spectrum of HD and contributing to the development of a practical regulatory strategy for disease-modifying (and potentially preventive) HD therapeutics.
FUTURE DIRECTIONS
In 2021, C-Path initiated an optimization to the HD-RSC to mirror other C-Path consortia. In 2022, the HD-RSC will be funded by consortium member organizations and governed by the C-Path Executive Director and an appointed industry co-director. The Coordinating Committee, consisting of representatives from each of the member organizations, plus scientific advisors who participate independent of their respective organizations, will convene to appoint consortium leadership (e.g., the industry co-director and Working Group co-chairs), assess the progress of working groups, and oversee the consortium portfolio, through a majority voting process. Membership may vary annually as drug developers launch new HD programs and internally reprioritize their portfolios, and it is expected to relaunch with 16 member organizations.
The HD-RSC can accelerate the path to regulatory approval by identifying and facilitating data-sharing opportunities and other open-science practices, advancing biomarkers and COAs in clinical studies, and fully characterizing disease progression with a view to clinical trial optimization across the entire course of HD. This consortium brings together HD drug development stakeholders, including regulatory agencies, to accelerate HD therapeutic development. The implementation of HD CDISC standards enables data integration for modeling, which will inform the evaluation of biomarkers and COAs. These tools will deepen our understanding of HD progression and pathobiology to steer therapeutics towards earlier prevention and define reliable measures of meaningful change. Progress in the themes first identified at the initial HD-RSC launch meeting and emphasized during subsequent HD-RSC meetings will catalyze the changes needed to develop effective therapeutics.
Footnotes
ACKNOWLEDGMENTS
The authors acknowledge critical reviewers of this manuscript, which include Simon Noble, Michael Panzara, Sarah Tabrizi, Yashmin Karten, Jacqueline Major, Kristen Swingle, and Klaus Romero. The Huntington’s Disease Regulatory Science Consortium received funding from CHDI Foundation, Inc., a nonprofit biomedical research organization exclusively dedicated to collaboratively developing therapeutics for Huntington’s disease.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.