
Abstract
RNA-sequencing technology helps to consider the expression of thousands of genes, simultaneously. The large-scale gene expression data include a huge number of genes versus a few samples. Therefore, the algorithms that among huge number of unrelated genes can accurately detect genes associated with specific disease can be useful for experts in early detect and treat the disease.
A two-phase search algorithm is proposed in this paper to discover the biomarkers in the RNA-seq gene expression dataset for the prostate cancer diagnosis. After statistical noise removing from the original large-scale dataset, a multi-objective optimization process is proposed to select the best non-dominated subset of genes with the maximum classification accuracy and the minimum number of genes, simultaneously. Finally, the proposed cache-based modification of the sequential forward floating selection (CMSFFS) algorithm is applied to the selected subset of genes to discover the most discriminant genes.
The obtained results show that the proposed algorithm is able to achieve the classification accuracy, sensitivity and specificity of 100% in the large scale RNA-seq prostate cancer dataset by selecting only three biomarkers.
Keywords
Introduction
The prostate is a small gland in the male reproductive system. Prostate cancer, which is also known as carcinoma of the prostate, occurs when some of the cells of the prostate, are reproduced much faster than normal case. RNA-sequencing data are known as large-scale data. Scientists use the large-scale data to check the differences between the normal and abnormal cells in the human body. Because of the high cost of genetic tests, the number of samples compared to the number of extracted genes is very low in the large-scale data. This is the main reason for the poor performance of many reported gene selection techniques. It is of great importance to develop an algorithm for the accurate detection of the genes associated with prostate cancer among huge number of unrelated genes. A number of researches dealing with the gene selection process from the large-scale prostate cancer data have been reported as follows.
Chiang and Ho [1] have combined the rough-based feature selection method with the radial basis function (RBF) neural network for the classification of gene expression data. This method can find the relevant features without requiring the number of clusters to be known a priori and identify the centers that approximate to the correct ones. In this paper, the authors have been attempted to introduce a prediction scheme that combines the rough-based feature selection method with radial basis function neural network. A hybrid gene selection method (IG-SVM) has been proposed in [2] to select informative genes for cancer classification. IG is a filter method that can eliminate irrelevant features in high-dimensional gene expression data and the wrapper SVM method has been used to further eliminate redundant genes based on the genes selected by filters. Given the small size of the datasets tested, the method proposed in this study should be further validated in larger datasets. Chen et al. [3] have proposed a gene selection method based on clustering, in which dissimilarity measures have been obtained through kernel functions. This method searches for best weights of genes iteratively at the same time to optimize the clustering objective function. Adaptive distance has been used in the proposed process in order to learn the weights of genes during the clustering process. Sharma et al. have proposed a gene selection method to classify the microarray gene expression data [4]. At first, the genes have been initially divided into a number of subsets. Then, the informative smaller subset of genes is merged with another gene subset in order to update the gene subsets. This method is repeated until all subsets are merged into one informative subset. An evolutionary algorithm based on genetic algorithms and artificial intelligence (IDGA) has been introduced in [5] to identify predictive genes for cancer classification. At first, a filter method has been applied to reduce the dimensionality of feature space. Next, an integer-coded genetic algorithm with variable-length genotype, adaptive parameters and modified genetic operators has been proposed.
An efficacious hybrid feature (gene) selection algorithm has been proposed in this paper in order to select the sub-optimal subset of genes for the classification of the large-scale RNA-seq prostate cancer data. Heuristic optimization techniques cannot guarantee the global optimum; therefore usually sub-optimal results are obtained. The sub-optimal results obtained in this investigation are acceptable.
At first, the noisy data are omitted by applying a statistical method. The sub-optimal subset of genes is selected from the RNA-seq prostate cancer dataset using the introduced Shuffled Frog Leaping-based Multi-objective Optimization Algorithm (SFLMOA) by considering the two objectives in this paper, i.e. “minimum number of selected genes” and “maximum amount of classification accuracy”. Then, the final subset of genes is selected using a Cache-based Modification of the Sequential Floating Feature Selection (CMSFFS) algorithm.
A combination of the teaching learning-based optimization (TLBO) algorithm and the PSO algorithm has been proposed in our previous work [6] to detect the smallest subset of genes involved in breast cancer with the highest performance. It is noted that in our previous work, a one-phase gene selection method based on a combination of two metaheuristic optimization techniques, i.e. the fuzzy adaptive PSO and TLBO, has been proposed for the feature selection of the breast cancer dataset. In contrast, in this study, a hybrid search algorithm based on the SFLMOA and the CMSFFS algorithm has been proposed to identify the most relevant genes. In other words, the combination of the introduced SFLMOA and CMSFFS algorithm in this study can find the sub-optimal subset of genes in the RNA-seq prostate cancer dataset.
The paper is organized as follows. The proposed hybrid gene selection algorithm is described in Section 2. In Section 3, the results are presented. Finally, the discussion and conclusion have been provided in Sections 4 and 5, respectively.
Proposed hybrid gene selection algorithm
In order to effectively represent the processes of the proposed hybrid gene selection algorithm, each of the components has been separately described.
The support vector machine (SVM) classifier
Vapnik et al. [7] have proposed the support vector machine which is a very effective and important machine learning approach. A critical issue in the large-scale data analysis, such as RNA-seq gene expression data, is the low number of samples versus a huge number of features which is known as “curse of dimensionality”. Support vector machines (SVMs) can easily overcome the challenge of the curse of dimensionality [8]. SVM-based methods maximize the margin between the original training data and the separating hyper-plane. If possible, a linear discriminant function is created based on a small number of critical border samples from each class otherwise, in order to automatically map the training samples into a space with higher dimension, the “kernel” technique is applied [9]. The linear, polynomial, radial basis function (RBF) and sigmoid functions as four basic kernels in SVM are respectively expressed as follows.
A statistical pre-processing with the following equation is used to reduce the primary dimension of gene expression data by removing the noisy genes.
Eusuff et al. [10] have originally developed the Shuffled frog-leaping algorithm (SFLA) which is a memetic meta-heuristic optimization to find the global optimal solution. Participating of each member in previous experience of all other members is the main idea of the SFLA. In the SFLA, frogs are described as a memetic vector and are hosts for memes. Each meme consists of a number of memotypes which the memotypes are similar to a gene in a chromosome in a Genetic Algorithm (GA). The frogs can communicate with each other in order to improve their memes by infecting each other. Position of an individual frog is changed by improving memes and is moved toward a global solution [11].
At first, initial population containing P frogs is randomly generated in the SFLA and then the frogs are divided into m memeplexes according to their fitness. The division is done so that the first high ranked frog going to the first memeplex, second one going to the second memeplex, the k-th frog go to the k-th memeplex and the k + 1-th frog back to the first memeplex.
The position of the worst frog in each memeplex at the iteration t is changed as follows.
If this process produces a frog with better fitness function value, it replaces the worst frog. Otherwise, x best is replaced by x gbest in Equation (7) and the above process is repeated in which x gbest is the global best solution. If no improvement becomes possible in this case, the worst frog in each memeplex is updated with a new random one.
After finishing the local search processes in the m memeplexes, all of the solutions are reshuffled and then the SFL algorithm is returned to the stage of sorting and division of the frogs into m memeplexes. The local search and the shuffling processes continue until the termination criterion is met.
In order to overcome the problem of trapping in the local optimum solutions, a new mutated version of the shuffled frog-leaping algorithm has been proposed in this paper. The mutation method is a strong technique to reclaim the efficiency of the SFL algorithm. In the mutated SFL algorithm and in each iteration, four frogs are selected randomly from the current memeplex. The mutated frog (
The mutation parameters are selected randomly in each iteration. For greater effectiveness, four selected frogs should be as:
Finally, in order to produce a new improved solution (frog), the generated mutated frog (
It should be noted that the bounds for all elements in each new generated solution should be checked. If any element in each new generated solution exceeds its limitations, it should be replaced by its own upper or lower bound.
In order to use the SFL algorithm in binary mode, the following equation have been introduced in [11]. By using this equations, the variables of each frog has been considered to be either “0” or “1” in the binary SFL (BSFL) algorithm which “0” or “1” in the position of each variable represents the absence or the presence of the specified gene, respectively. The position vector (x) in the BSFL algorithm is updated by using the Δx
k
in each iteration.
Let [V
i
] be the set of i features from the original set [P]. The importance of the feature v in the set [V
i
] and the feature p in the set [P - V
i
] are respectively calculated using Equations (12 and 13).
In the first stage of the Cache-Based Modification of the Sequential Forward Floating Selection (CMSFFS) algorithm, the Sequential Forward Selection (SFS) method [12] has been used to select the initial subset of features. In the CMSFFS algorithm, the search process has been started from the initial subset of features ([V2]). In the first stage, the most important member (p) from the [P - V i ] set is added to the subset of [V i ] and generate [Vi+1] if the candidate feature is not listed in the Black List and the subset of [V i ] + p is not listed in the Cache cell.
If [Vi+1] is in the Cache memory, the most important member is added to the Black List and the process to select the most important member for aggregating with [V i ] is repeated.
Let v l be the least important feature in [Vi+1]. In this case, if v l = p then the new generated [Vi+1] is stored in the Cache memory, i + 1 is considered as i and the process of the most important member selection to be aggregated with [V i ] is repeated after clearing the Black List. If v l ≠ p then v l is deleted from [Vi+1], and [V′ i ] is generated and the second stage of the proposed CMSFFS algorithm is started.
In the second stage, if the number of members of [V′ i ] is equal to the number of members of [V initial ], [V i ] is replaced by [V′ i ], CF ([V i ]) is replaced by CF ([V′ i ]), the new generated [V′ i ] is stored in the Cache memory and the process of the most important member selection to be added to [V i ] is repeated after clearing the Black List. Otherwise the least important feature in [V′ i ] is detected as v ll . If CF ([V′ i ] - v ll ) ≤ CF ([Vi-1]) then [V i ] is replaced by [V′ i ], CF ([V i ]) is replaced by CF ([V′ i ]), the new generated [V′ i ] is stored in the Cache memory and the process of the most important member selection to be combined with [V i ] is repeated after clearing the Black List. Otherwise v ll is deleted from [V′ i ] and [V′i-1] is generated then i - 1 is considered as i. Then the condition of equality of the number of members of [V′ i ] and the number of members of [V initial ] is checked again. This process continues until the desired number of features achieves. More details of the CMSFFS algorithm are available in [13].
In order to resolve the weakness of some previous works [4, 14– 20] (considering the feature selection problem to be single objective), the optimization problem has been considered bi-objective in this paper. In this case, the highest classification accuracy and the smallest subset of genes have been simultaneously considered by employing a multi-objective approach and storing the non-dominated solutions in the matrix repository (Rep). Details of the multi-objective technique used in this paper are available in [21].
Assuming the minimization problem, the concept of the non-dominated solution is described as follows:
If x1 and x2 are two candidate solutions in the multi-objective problem and x1 and x2, then the following relation must be satisfied.
If f1 and f2 are, respectively, the objective functions of classification accuracy and the number of selected genes, their membership functions are defined as follows.
f1 and f2 are, respectively, in the range of 0 ≤ f1 ≤ 1 and 1 ≤ f2 ≤ m that m is the largest number of genes considered in the optimization process.
Since the optimization problem in this study is in a multi-objective form, the direct calculation of x
worst
, x
best
and x
gbest
(in the SFL algorithm) is not possible. In this study, to choose the best solutions for x
best
and x
gbest
at each iteration of the proposed SFLA-based multi-objective optimization algorithm, a fuzzy interactive (min-max) method has been considered according to the goal defined in Equation (17). Also, in order to choose the worst solution (x
worst
) at each iteration of the proposed SFLA-based multi-objective optimization algorithm, the max-max method has been proposed according to the goal defined in Equation (18).
Figure 1 represents the flowchart of the proposed gene selection algorithm in this study.
In this study, an approach based on the shuffled frog-leaping algorithm (SFLA) and the CMSFFS algorithm is proposed to identify the most informative genes in RNA-seq prostate cancer dataset. In the first stage, the mutated binary SFLA-based multi-objective optimization is used to simultaneous incorporate both objectives of the maximum accuracy and the minimum number of selected genes in the proposed gene selection algorithm. Finally, the cache-based modification of the SFFS algorithm is used to choose the most discriminant genes from those selected in the SFLA-based multi-objective optimization stage. The SVM classifier with RBF kernel has been considered as the central core of the proposed approach to classify the RNA-seq prostate cancer data.
Let
If
Experimental results
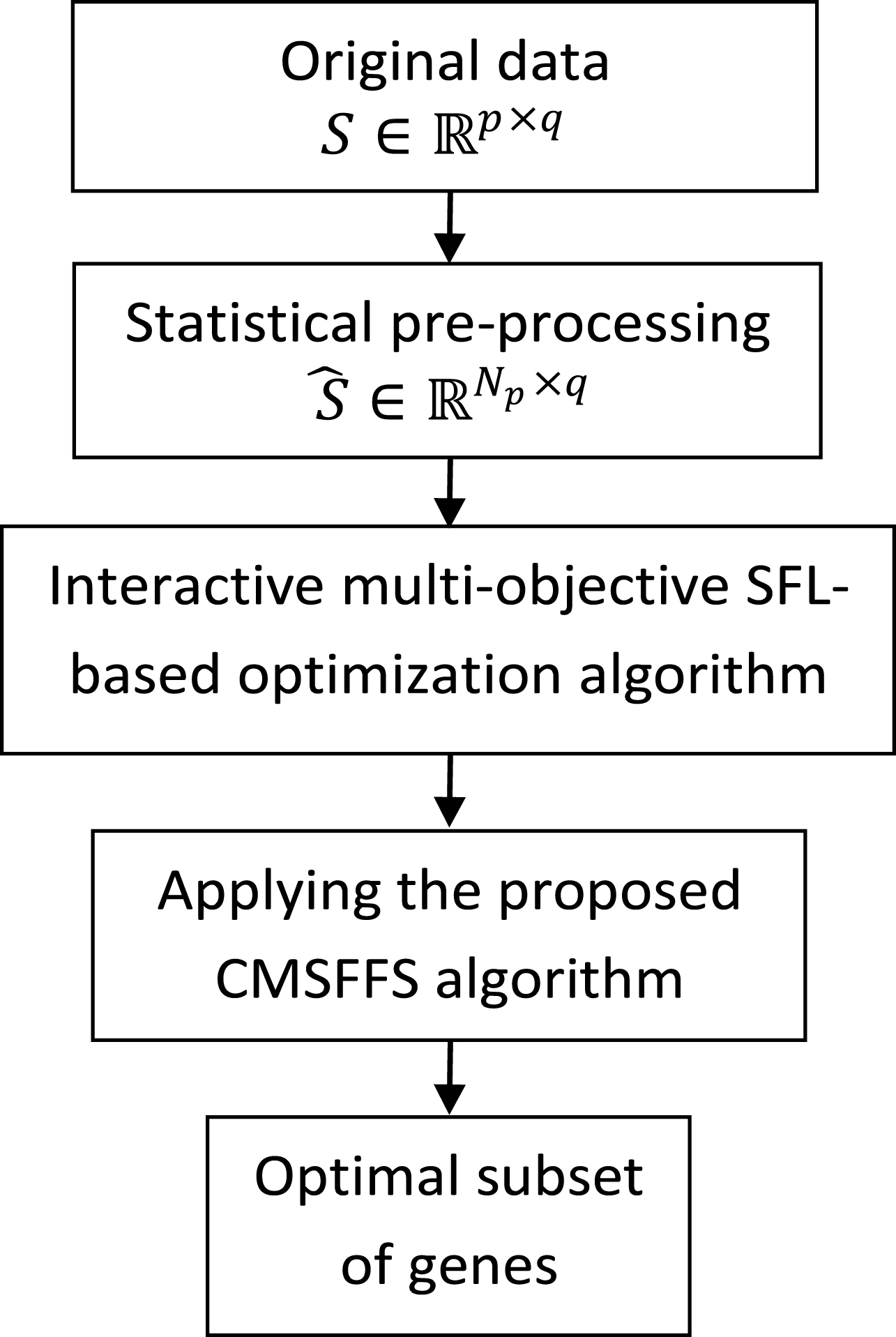
Flowchart of the proposed gene selection algorithm.
In order to evaluate the performance of the proposed gene selection algorithm in this paper, the publicly available RNA-seq prostate cancer dataset [22] has been employed. The RNA-sequencing prostate cancer dataset consists of 20502 genes and 20 samples with 10 benign prostate tissues and 10 cancerous prostate tissues. Also, the number of classes in these datasets is two.
Assessment process
In order to prevent the over-fitting and leaking phenomenon, 20% of the samples in the RNA-seq prostate cancer dataset is separated as external test data after shuffling the original data by applying the five-fold cross-validation in the gene selection process; the remaining 80% of the samples is only used in the proposed gene selection process. Therefore, the external test data mentioned above are used to evaluate the results obtained in the process of the proposed gene selection algorithm. Hence, the final results reported in Table 1 are based on the external test data. Therefore, it can be claimed that only the external test data, which have never participated in the gene selection process, have been used to evaluate the final results of the proposed gene selection algorithm.
Comparison of gene selection algorithms from the large-scale prostate cancer datasets
Comparison of gene selection algorithms from the large-scale prostate cancer datasets
*N/A stands for not available.
By considering the two classes of “normal” and “tumor” in the RNA-seq prostate cancer gene expression dataset, the following parameters are defined to evaluate the performance of the proposed gene selection algorithm. TP (True Positive) represents the samples which algorithm TN (True Negative) represents the samples which algorithm FP (False Positive) represents the samples which algorithm FN (False Negative) represents the samples which algorithm
In this study, the ratio of tumor cases that the classifier correctly classifies them in the tumor class is known as the sensitivity. The ratio of normal cases that the classifier correctly classifies them in the normal class is known as the specificity. Accuracy is another important quantitative criterion which represents the number of samples that the proposed gene selection algorithm diagnoses their affiliation or non-affiliation to the specified group. Three explained criteria are mathematically formulated as follows.
As explained before, in order to remove the noisy genes and to select the most informative genes in the RNA-seq prostate cancer dataset to be sent to the mutated SFLA-based multi-objective optimization stage, the statistical pre-processing has been used. Figure 2 shows the result of applying the pre-processing stage on the RNA-seq prostate cancer dataset. In this figure, the horizontal axis and the vertical axis respectively indicate the number of selected genes with the highest scores and the average of classification accuracy. The best value for N p to be sent to the proposed multi-objective stage has been considered N p = 100. It can be seen that for N p = 100, there is a flat area in Fig. 2 in which by increasing the number of genes, the maximum classification accuracy is not affected.

The result of applying the pre-processing stage on the RNA-seq prostate cancer dataset.

The result of applying the proposed multi-objective optimization method on the RNA-seq prostate cancer dataset.
In this study, the number of initial population in the proposed multi-objective optimization stage has been considered N = 300.
As mentioned before, the binary SFL algorithm has high capability to find the sub-optimal solution. Also, the mutation method used in the SFL algorithm increases the capability of local search of the proposed optimization algorithm. The proposed mutated SFL-based multi-objective optimization method has been applied to the filtered RNA-seq data in order to select the sub-optimal subset of genes by considering, simultaneously, the two defined objectives, i.e. “minimum number of selected genes” and “maximum amount of classification accuracy”. In this paper N I = 10 has been considered. Applying data folding technique with N F = 5 and also the repetition of the validation process in the proposed SFLA-based multi-objective optimization algorithm with N R = 20 enhance the robustness and reduce the randomness of the proposed algorithm.
Figure 3 demonstrates the result of applying the first stage of the gene selection process, i.e. the proposed multi-objective optimization method on the RNA-seq prostate cancer dataset. In Fig. 3, the horizontal and the vertical axes specify the accuracy of the RNA-seq data classification and the number of selected genes, respectively. The star-shaped symbols on the pareto-front indicate the sub-optimal solutions which the sub-optimal solution with the highest classification accuracy and the lowest number of genes has been marked in Fig. 3. As indicated in Fig. 3, the proposed mutated SFL-based multi-objective optimization method is able to achieve the classification accuracy of 100% in the first stage of the gene selection process with only nine genes.
In the second stage of the proposed gene selection approach, the CMSFFS algorithm has been applied to the sub-optimal subset of genes selected in the multi-objective optimization stage.
By applying the CMSFFS algorithm, the final subset of genes is selected from the RNA-seq prostate cancer dataset in a repetitive cache-based process. The final result of applying the CMSFFS gene selection algorithm on the sub-optimal subset of genes selected in the multi-objective optimization stage on the RNA-seq prostate cancer dataset has been shown in Fig. 4.
The horizontal and the vertical axes in Fig. 4 represent the number of selected genes and the classification accuracy of the RNA-seq prostate cancer data. As shown in Fig. 4, the proposed gene selection algorithm is capable to select the sub-optimal subset of genes with only three genes in the large scale RNA-seq prostate cancer dataset such that 100% classification accuracy can be achieved.

The final result of applying the CMSFFS gene selection algorithm on the sub-optimal subset of genes selected in the multi-objective optimization stage on the RNA-seq prostate cancer dataset.
Table 1 represents a comparison of the results of the mutated SFL-based gene selection algorithm proposed in this paper and several researches in recent years on the large-scale prostate cancer datasets.
A combination of the rough-based feature selection method and RBF neural network has been used in [1] in order to classify the gene expression data. The number of folds has been considered to be 10 and the accuracy value of the classification has been reported 99.62% with the 668 genes selected from the prostate cancer dataset. In [2], A hybrid SVM-based gene selection method has been used to analysis the gene expression data. The accuracy of the classification in [2] has been obtained 96.08% with 3 selected genes. It should be noted that the folding technique with the number of folds of 10 has been used in this study. A clustering-based gene selection method has been proposed in [3]. The number of folds in this study has been considered 10 for the prostate cancer dataset and the accuracy of the classification in [3] has been obtained 96.85% with 10 selected genes. In [4], a feature selection algorithm has been proposed based on the division of genes into small subsets which the subsets of features provide high classification accuracy. The number of folds has been considered to be 3 for the prostate cancer dataset. The accuracy of the classification in [4] has been obtained 97% with 4 selected genes. A method based on genetic algorithm has been proposed in [5] to select the informative genes for cancer classification. The accuracy value of the prostate cancer data classification has been obtained 96.3% while the number of selected genes has been reported 14. Also, the number of folds has been considered to be 10.
Most of the techniques proposed in recent years could optimize only one of the two important objectives, i.e. minimization of the number of selected genes or maximization of the classification accuracy in the considered gene expression dataset.
In this study, at first the noisy genes are filtered in the statistical pre-processing stage. Then by applying the proposed fuzzy interactive multi-objective SFL-based optimization algorithm on the filtered RNA-seq prostate cancer dataset, the sub-optimal subset of informative genes associated with prostate cancer is obtained. In the proposed binary SFL-based multi-objective optimization algorithm, the ability of local search has been increased by employing the proposed mutation method with 5 members (x gbest and 4 different members) for each member of the population. In the multi-objective optimization stage, the sub-optimal subset with the lowest number of genes and the highest classification accuracy is selected. Our proposed SFL-based multi-objective optimization method can achieve 100% accuracy of classification by selecting 9 genes of the RNA-seq prostate cancer dataset. In the second stage of the proposed search approach, the sub-optimal subset of genes obtained from the multi-objective optimization stage is sent to the CMSFFS algorithm. Ultimately, the most informative genes are selected from the sub-optimal subset of genes by applying the CMSFFS algorithm.
The final subset of genes with only the three genes has been selected by applying the proposed hybrid gene selection approach on the RNA-seq prostate cancer dataset so that 100% accuracy of classification has been achieved.
By using the folding technique and repeating the assessment process several times, the phenomenon of randomness is reduced in this study and the robustness of the proposed algorithm is increased. In this paper, to evaluate the proposed algorithm, the SVM classifier has been used because the SVM classifier can overcome the challenge of “curse of dimensionality”. Because of the high dimensionality of the search space in the RNA-seq prostate cancer dataset, the proposed hybrid gene selection approach could find the global sub-optimal solution in the two proposed stage.
Details of the sub-optimally selected genes in the RNA-seq prostate cancer dataset has been listed in Table 2.
Details of the final subset of genes in the RNA-seq prostate cancer dataset
Details of the final subset of genes in the RNA-seq prostate cancer dataset
A powerful hybrid gene selection algorithm based on multi-objective optimization was suggested in this paper to select the most informative subset of genes in the RNA-seq prostate cancer dataset. After removing the noisy genes, the sub-optimal subset of genes associated with prostate cancer was selected in the multi-objective optimization stage. The combination of the proposed mutation method by the SFL-based optimization algorithm in binary mode could reduce the probability of being caught in the local optimum and increase the ability of local searching of the proposed algorithm. Using multi-objective optimization process in this article could enable the decision maker to select the required solution according to the number of genes and the classification accuracy. Ultimately, in order to detect the final subset of genes from the RNA-seq prostate cancer dataset, the CMSFFS algorithm was used. The final subset of genes with the three identified genes could classify the RNA-seq data with the classification accuracy, sensitivity and specificity of 100% by using the external test data. Also, the probability of obtaining random results was minimized in this study by using the techniques of data folding and multiple implementation of the proposed gene selection algorithm.