
Abstract
Chromosome visualization has been used in human chromosome analysis and is a crucial step in clinical diagnosis and drug development. An important step in chromosome visualization is the extraction of chromosomes from chromosome images obtained by light microscopy. Chromosomes often overlap in a complex and variable manner, resulting in significant challenges in chromosome segmentation. The process of chromosome visualization requires manual intervention and is tedious. A method based on a neural network is proposed for the automatic segmentation of overlapping chromosome images to speed up the workflow of visualizing chromosomes. Three improved dilated convolutions are used in the chromosome image segmentation models based on U-Net. The proposed models successfully segment overlapping chromosomes in two publicly available overlapping chromosome data sets. Our models have better performance than existing overlapping chromosome segmentation methods based on U-Net. In summary, it is demonstrated that the improved dilated convolutions can be used for the automatic segmentation of overlapping chromosome images. The proposed improved dilated convolutions have a stable performance improvement, can be easily extended to the segmentation of multiple overlapping chromosomes, and are suitable as general neural network operations to replace standard convolutions in any network.
Keywords
Introduction
Chromosomal abnormalities can lead to birth defects. At least 8 million new birth defects occur every year worldwide, accounting for about 6% of all births. The diseases caused by chromosomal abnormality include malformation, mental retardation, sexual development abnormality, and some cancers, which affect human health and population quality [3]. Nowadays, there is a lack of drugs and treatments for chromosomal diseases; thus, it is necessary to perform chromosomal examination and analysis of patients to provide a scientific basis for prenatal diagnosis, eugenics, and drug development.
A chromosome is a thread-like structure carrying the human genetic material DNA. The structure of chromosomes is complex and subtle and includes a short arm, long arm, satellite, euchromatic region, primary constriction, secondary constriction, heterochromatic region, and special telomeres at the end of the chromosomes. There are 46 chromosomes in normal human somatic cells, including 22 pairs of autosomes and 2 sex chromosomes [1]. Genes are DNA fragments that contain genetic information. Chromosomes are carriers of DNA; therefore, abnormalities in their number or structure can lead to genetic defects. These abnormalities may be inherited from parents or may be due to mutations.
Fluorescence in situ hybridization (FISH) is an important genetic experimental technique for detecting chromosomal and genetic abnormalities [2]. According to the principle of complementary base pairing, a probe with a fluorescent substance is conjugated with the target DNA by special means so that the position of the target DNA can be observed by a fluorescence microscope. FISH can be used to detect the structural variation of chromosomes and detect missing, additional, or replaced chromosomes easily. Image processing of FISH is usually conducted by cytogeneticists in gene laboratories, including the collection of chromosome images from a microscope, the segmentation of chromosomes in the images, and the arrangement of individual chromosomes in a standard format. This process is time-consuming and laborious, requires numerous professional and technical personnel, and often results in errors due to differences in the processing quality among the personnel and fatigue. The segmentation of overlapping chromosomes is the most difficult task in chromosome image processing because it depends entirely on manual processing. Therefore, the automatic segmentation of chromosomes is highly desirable.
Chromosome overlap has been researched for more than 50 years [3]. With the development of computer software and hardware, computer technology has been increasingly used for chromosome analysis [4]. Based on digital image processing, topology, and mathematical methods, researchers have integrated the use of gray-level information, contours, and texture information for the segmentation of chromosomes. These studies include threshold-based segmentation [5–7], Fourier transform [8], Delaunay triangulations, Voronoi diagrams [9], watershed-based segmentation [10, 11], maximum likelihood methods [2, 12], level set evolution [13, 14], and fuzzy c-means clustering [11, 15].
However, a major challenge with chromosomes is that they change their appearance during the cell cycle. Mitosis in the cell cycle includes four stages: prophase, metaphase, anaphase, and telophase. The structure of chromosomes can only be clearly observed in the metaphase [16]. Moreover, chromosomes are not rigid. They can twist, flip, and bend at different angles, and unfortunately, this occurs in almost every cell image in the metaphase, impeding the ability to distinguish chromosomes using a microscope. In addition, the quality of chromosome images largely depends on the illumination, spatial projection, sample preparation quality, and image acquisition equipment. Therefore, many problems are encountered in chromosome micrographs, such as impurity, blur, touching, and overlap, complicating the characterization of chromosome features.
Semantic segmentation based on a convolutional neural network (CNN) has great potential for overcoming these challenges. This approach is a research direction in the field of artificial intelligence and machine learning. A CNN is a deep learning method capable of automatically obtaining features from input images, without a need to extract features manually. The latest development in computer vision and image segmentation is remarkable [17]. Some researchers have confirmed the excellent performance of CNNs for image processing of electron microscope images [18, 19]. Deformable segmentation via sparse representation and dictionary learning has also yielded remarkable results [46–48]. Ref. [49] used the attention mechanism in the expansion path of U-Net for retinal vessel image segmentation, whereas we use improved dilated convolutions in the U-Net contraction path to improve the performance of chromosome image segmentation. A combination of the two methods could be investigated in a future study.
Neural network-based automated systems for human chromosome classification and karyotype detection were investigated [20]. Crowdsourcing and CNN-based automatic segmentation models have been used to segment and classify overlapping chromosomes [21]. Ref. [22] proposed an end-to-end chromosome classification and segmentation method with data segmentation using a Multiple Distribution Generative Advertising Network (MD-GAN). A U-Net-based CNN was customized for the segmentation of overlapping chromosomes; the output feature map had the same size as the input image [23]. Ref. [24] improved the network design for chromosome segmentation tasks by determining the appropriate number of U-Net layers and test time augmentation. However, existing CNN-based chromosome segmentation models all use small filter kernels (3×3 or 1×1). Although it is highly effective to obtain long-range information on deep blocks through concatenated convolutions with small kernels in classification tasks, large kernels with large receptive fields have provided good results in the field of semantic segmentation. The reason is that in semantic segmentation, classification and location tasks are executed simultaneously to make dense predictions for each pixel. That is to say, it is more advantageous in semantic segmentation to use kernels with large receptive fields to obtain large-scale coding information in the same layer.
Although large kernels are very useful for semantic segmentation, standard large convolution kernels will increase the computational complexity, which is not conducive to an increase in the depth of the model and may reduce the model performance. In the case of a small amount of data, it will lead to overfitting. Unfortunately, chromosome training data are often difficult to obtain because specimen preparation, image acquisition, and labeling require time, human resources, and expertise.
Dilated convolution [25] performs very well for semantic segmentation tasks [26–28]. Dilated convolution, also known as atrous convolution, is equivalent to inserting holes between the weights of filters of standard convolution. Dilated convolution is an ingenious method for CNNs to perform multi-scale processing and uses filters with different dilation rates to extract multi-scale information. In our work, a small filter (3×3) commonly used in standard convolution is applied to obtain short-range information and dilated convolution to obtain long-range information. This strategy makes use of multi-scale context information without adding extra parameters and allows for a powerful segmentation method for overlapping chromosomes using a small set of training data.
Recently, dilated convolution has been widely used in the localization and segmentation of prostate cancer [29], rectal cancer [30], lung disease [31], intervertebral disc [32], brain tumor [33], tongue contours [34], and breast cancer [35]. As far as we know, this is the first time that dilated convolution has been used for overlapping chromosome segmentation.
Here, the applicability of dilated convolution for the automatic segmentation of overlapping chromosomes is investigated. The task is achieved by incorporating improved dilated convolutions into the U-Net network structure. First, existing smoothed dilated convolution with separable and shared (SS) convolution and smoothed dilated convolution with a group interaction (GI) operation in U-Net are used. Subsequently, dilated convolutions with group shared (GS), full information (FI), and full information-group interaction (FIGI) operations are applied in the chromosome segmentation models. The results of the proposed model are compared with those of the two latest models on two publicly available chromosome datasets to verify the performance of the proposed segmentation model based on the improved dilated convolutions. The experimental results show that dilated convolution provides excellent performance for overlapping chromosome segmentation. The three proposed improved dilated convolutions provide better performance than comparable methods and have the potential to become general neural network operations.
Section 2 describes the materials, the network architecture, the three existing convolution operations used in this work, the three proposed convolution operations, and the performance metrics. Section 3 summarizes the results and provides the discussion, and Section 4 concludes with suggestions for future work.
Materials and methods
FISH specimen preparation
The Q-FISH method, which uses fluorescent molecules to stain genes or chromosomes, is particularly suitable for gene location and chromosome abnormality recognition [36]. This method involves adding colcemid to the culture medium to disrupt the microtubules in the mitotic cells and arrest the cells in the metaphase state. Next, the cells are trypsinized and suspended in a hypotonic buffer, causing the cells to break down and the chromosomes to spread. Subsequently, the solution is removed by centrifugation and resuspended in methanol/glacial acetic acid. Drops of the cell suspension are placed on a microscope slide, and the slide is dried overnight and immersed in phosphate buffer saline (PBS) for a few minutes. The slide is then fixed in a 4% formaldehyde solution for a few minutes, followed by PBS washing. The slide is placed in a pepsin solution to digest the proteins. A small amount of the hybrid mixture is placed on a cover slide, which is placed on the prepared slide. The slide is then placed in an oven at 80°C to denature the chromosomal DNA. The slide is kept at room temperature for several hours to hybridize the probe with complementary DNA. Subsequently, the slide is washed with a wash solution to remove the unbound probe. The microscope mounting medium, which contains a DNA counterstain and an antifade solution, is placed on the slide.
Data acquisition and processing
FISH images of the chromosomes in the cells were taken from the chromosome specimens fixed on the slides. The FISH images are 12-bit gray-scale TIF files, with a size of 1536×1024 pixels. The images of the metaphase cells contain two images taken at two wavelengths (blue and red) and showing the human metaphasic chromosomes (DAPI stained) hybridized with a Cy3-labeled telomeric probe, namely a chromosome image and a telomere image. One or more chromosomes in a chromosome specimen on a slide appear on top of another chromosome, resulting in overlapping chromosomes in the image. The challenge is that different types of chromosomes can overlap at different angles and relative positions, making detection difficult using geometric-based methods. Therefore, a deep neural network is used to learn the features of the overlapping chromosomes from the training data sets. Screening in the metaphase cells is too time-consuming and laborious to obtain enough overlapping samples; therefore, the data set is generated by systematically combining individual chromosomes (Fig. 1).

Overlapping chromosomes and corresponding ground truth maps in the dataset. The images in the odd columns show overlapping chromosomes, and the images in the even columns show the corresponding ground truth maps. This dataset contains X-type, T-type, and other types of overlapping chromosome images.
The datasets used in this work are publicly available [37, 38]. The preparation procedure of the dataset was as follows: firstly, 12-bit gray-scale images of the chromosomes and telomeres obtained from the microscope were converted into 8-bit gray-scale images to facilitate the segmentation and generation of RGB images. The gray-scale images of the chromosomes and telomeres were processed using adaptive threshold and morphological methods. Then the processed chromosome image and telomere image were combined. Morphological processing was used to extract all single chromosomes, and training labels were generated. The chromosomes and labels were combined into 2-channel images; 1,035 possible chromosome pairs were generated as a result of the Cartesian product of the 2-channel images.
Each chromosome was rotated by a specific angle, and each pair one chromosomes was translated horizontally and vertically generate a large number of chromosome overlaps. The problem is that this method way of generating chromosome overlaps results in a large dataset that may exceed the RAM capacity. In this case, it is preferred to use many different chromosome pairs than to apply many rotations and translations to the same chromosome pair. Methods to achieve this include reducing image resolution and the number of rotations and translations. These methods result in a larger number of chromosome pairs while reducing the need for more RAM.
First, a basic network (U-Net-basic) was created similar to the U-Net developed by Hu et al. [23]. Improved dilated convolutions were combined with U-Net-basic to achieve accurate segmentation of the overlapping chromosomes. U-Net-basic consists of a contraction path (encoder) and an expansion path (decoder), but it is not symmetrical. The size of the chromosome image, batch size, the number of network layers, the size of the filters, the number of skip connections, and the location of the dilated convolution were determined through experimental methods to minimize the computing resources. The optimum results were obtained by setting the filter size of the standard convolutions to 3×3, the filter size of the transposed convolutions to 2×2, the stride to 2, and the maximum pooling size to 2×2. The input chromosome images of the first convolution layer were resized to 80×80. The first block contained three standard convolutions, each of which had 64 filters; this was followed by maximum pooling. The second block used the output feature map of the first block as an input. It had 2 standard convolutions with 128 filters and maximum pooling. The third block used the output of the second block as input and had two standard convolutions with 256 filters, followed by a transposed convolution with 128 filters. The output of the third block and the output of the second block were concatenated and became the input of the fourth block. The fourth block contained two standard convolutions and one transposed convolution. The number of filters of convolutions and transposed convolutions was 128 and 64, respectively. The output of the fourth block and the output of the first block were concatenated and became the input of the fifth block. The fifth block contained two standard convolutions of 3×3 and one convolution of 1×1. The number of filters of the 3×3 standard convolution and 1×1 convolution was 64 and 4, respectively. The first block and the second block comprised the contraction path (encoder), and the fourth block and the fifth block represented the expansion path (decoder).
All subsequent models were improved based on the basic model. As shown in Fig. 2, the models had a similar network structure, which included improved dilated convolutions in the first three blocks. The differences between the models were the improved dilated convolutions. The number of improved dilated convolutions that were added and the locations were determined experimentally.
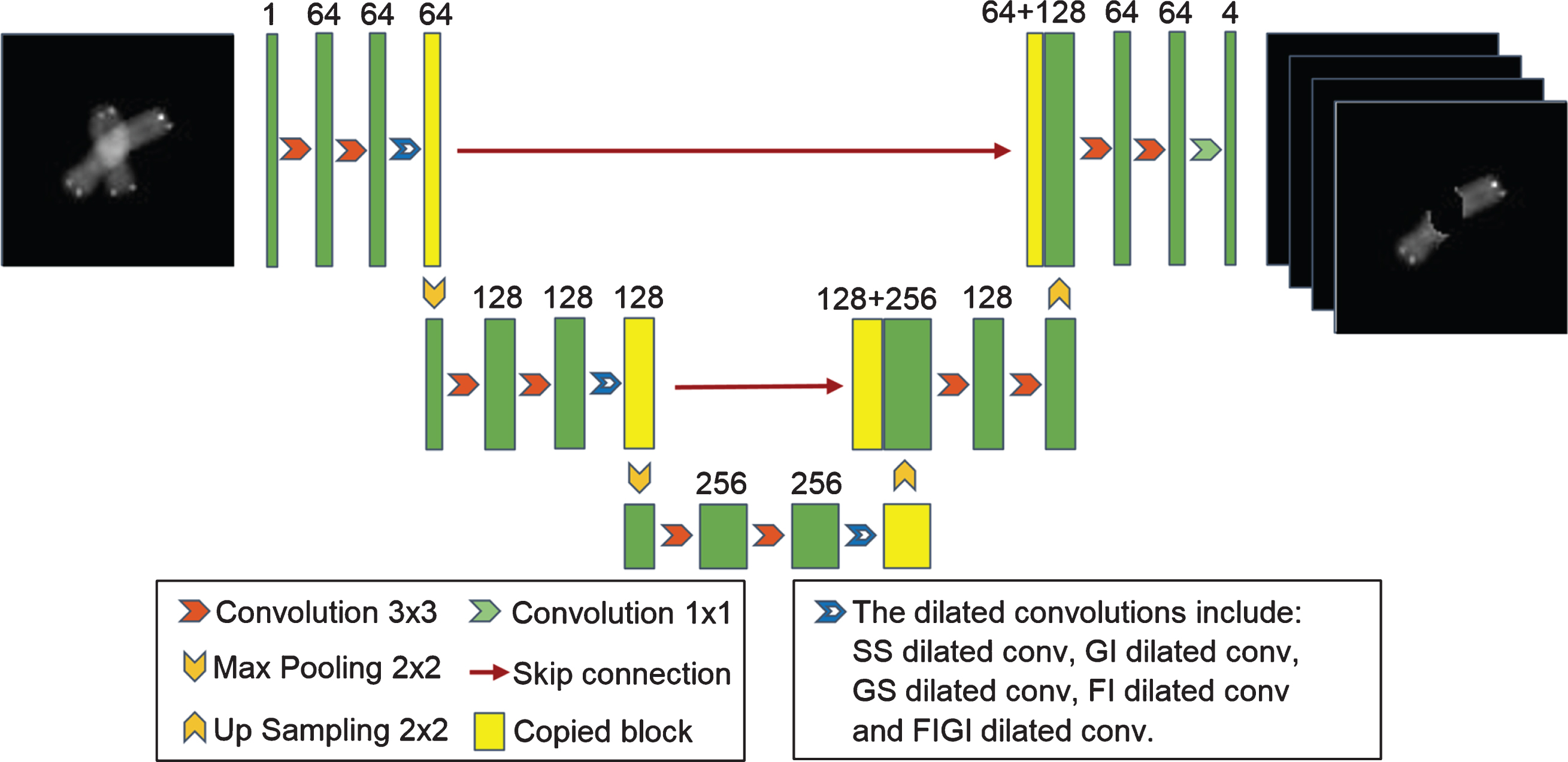
Architecture of the network for the segmentation of overlapping chromosome images. Each green box corresponds to a multi-channel feature map. The number of channels is shown above the box. The dilated convolutions are added to the blocks of the contraction path. The yellow boxes represent the copied feature maps. The arrows denote the different operations. The dilated convolutions include SS dilated conv, GI dilated conv, GS dilated conv, FI dilated conv, and FIGI dilated conv. The details are provided in Sections 2.5–2.9.
Dilated convolution, also known as atrous convolution or convolution with holes, consists of adding holes to the standard convolution kernel to increase the reception field [39]. Compared with the standard convolution, the dilated convolution has an extra hyperparameter called the dilation rate, which represents the number of intervals in the kernel. For example, the dilation rate of a standard convolution is 1. By setting different dilation rates, the size of the region observed by the same dilated convolution kernel can be changed, whereas the standard convolution does not have this flexibility. Dilated convolution enlarges the receptive field exponentially without losing resolution or coverage.
The improved dilated convolutions were based on a decomposition of the dilated convolution [28]. Dilated convolution is equivalent to a set of operations, including decomposition, convolution, and recombination. This set of operations is executed as follows. First, an input feature map is deinterlaced to feature maps of reduced resolution by periodic subsampling. Second, all reduced resolution feature maps undergo the same standard convolution, which has filters with the same weights as the original dilated convolution after removing all the holes, i.e., the standard convolution is the same for all the reduced resolution feature maps. The final step is to re-interlace the feature maps to produce outputs with the original resolution. This process transforms dilated convolutions into standard convolutions and results in high efficiency [40–42].
In this work, an existing smoothed dilated convolution with an SS operation (SS dilated convolution) [28] and a smoothed dilated convolution with a GI operation (GI dilated convolution) [28] were implemented. Additionally, three improved dilated convolutions with GS, FI, and FIGI operations (GS dilated convolution, FI dilated convolution, and FIGI dilated convolution) were used. The dilated convolutions were used to establish models with the structure shown in Fig. 2.
SS dilated convolution
The SS dilated convolutions were added to the contraction path of U-Net-basic to obtain large-scale coding information (Fig. 2); this new model is referred to as U-Net-SS. Smoothed dilated convolution is an improved method of dilated convolution that can partially overcome the gridding artifacts caused by the loss of local dependency information in dilated convolution [28]. The method is equivalent to inserting a SS operation before a dilated convolution. The SS operation adds intergroup dependency information before the convolutions in the second step of the decomposition. Since the number of channels does not increase, there is mutual interference between group dependence and information within the group.
GI dilated convolution
GI dilated convolution is another improved method of dilated convolution proposed by [28]. The GI operation is similar to a fully connected layer, except that the operation targets are groups. The operation rebuilds dependencies among different groups in the third step of the decomposition. It was implemented and added to the basic network architecture (Fig. 2). When the dilation rate is r, and the input feature map dimension is d, the operation produces rd groups of feature maps of reduced resolution in the second step of the decomposition. A GI layer has a weight matrix
GS dilated convolution
As mentioned above, the decomposition operation of dilated convolution disperses the adjacent units in the input feature map to different reduced resolution feature maps, i.e., the adjacent units in a reduced resolution feature map are not neighbors in the input feature map. Local dependency information is in this periodic sampling, which will lead to gridding artifacts. If local dependency information is added to each unit of the input feature map before the periodic subsampling process, the gridding artifacts will be reduced.
The GS dilated convolution is shown in Fig. 3. Before the first step of the decomposition of dilated convolution, the GS convolution operation is added to smooth the input feature map so that local dependency information is added. In this way, the local dependency information is obtained by intermediate feature maps during periodic subsampling and is used by subsequent standard convolutions to improve the results.
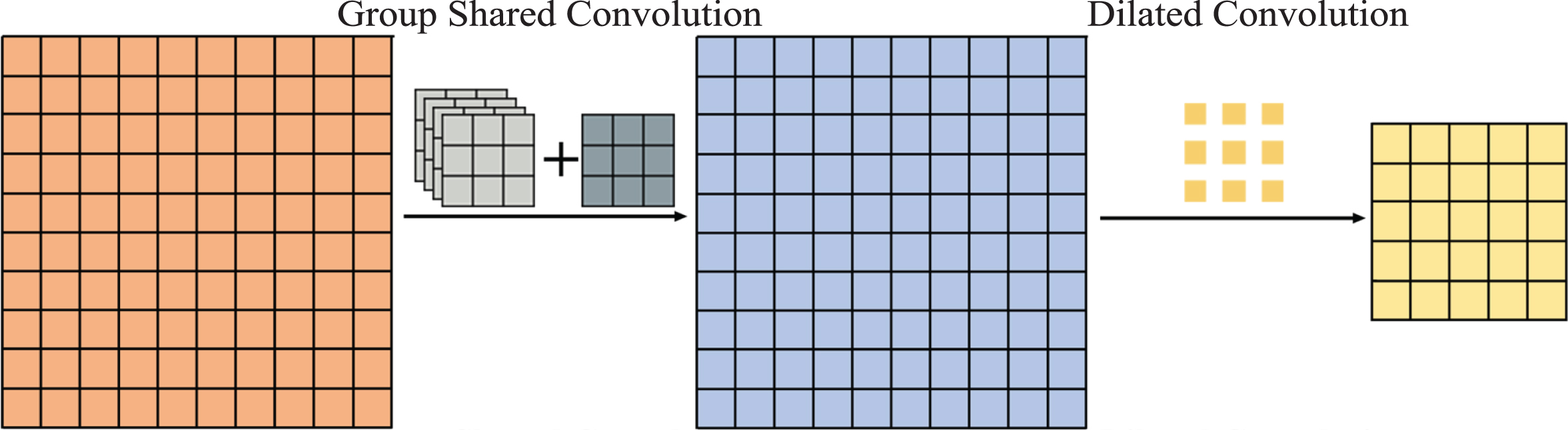
Dilated convolution with a group shared convolution operation, which eliminates gridding artifacts. Group shared convolution is used to add local dependency information to the input feature map before the periodic subsampling of dilated convolution.
GS convolution is an improvement of the SS convolution [28] and separable convolution [27, 44], and the SS convolution is an improvement of the separable convolution. The convolution kernels of the separable convolution are different for multiple channels; therefore, the local dependency information they extract depends on the channel. Although this information represents the inherent differences between channels, the method is not very compatible with dilated convolution and cannot be used before a dilated convolution to add local dependency information. Multiple channels of the convolution kernel of the SS convolution share parameters, which solves the problem of the separable convolution. However, because SS convolution shares the same convolution kernel for the smoothing operation on all channels to obtain local dependency, the information of the channels is greatly reduced.
Therefore, GS convolution is proposed to minimize the reduction of channel information. Due to the diversity of local information among different channels, the same shared convolution is not directly applied to all channels. First, the channels are grouped, and each group of channels shares a convolution kernel. Then the results are concatenated and convolved with a shared convolution kernel. The smoothing operation is gradually completed using these two steps.
Compared with SS convolution, GS convolution has additional channel groups, and the channels in each group share the convolution kernel. For a dilated convolution with a dilation rate of r and feature maps with dimension d and channel number C, GS convolution is defined as follows. First, a feature map of C channels is divided into four small feature maps based on the number of channels, and the number of channels of each small feature map is C/4. The channels of each small feature map share a convolution kernel; thus, four intermediate feature maps are generated. Subsequently, the four intermediate feature maps are concatenated into a feature map with the same channel numbers as the input. Finally, all the channels of the feature map are convolved with a shared convolution kernel.
When the input feature map has only one channel, the GS convolution and the SS convolution are identical. However, when the channel number C > 1, the GS convolution will make better use of the local dependency information of multiple channels, and only 4 (2r - 1) d parameters will be added. This means that the proposed method uses 5 (2r - 1) d parameters in total, and the number of parameters does not increase with the number of channels. In practice, the number of parameters is small. After adding two dilated convolutions with a GS convolution operation to U-Net-basic, a model called U-Net-GS was created.
In SS convolution and GS convolution, the number of channels in the feature map does not increase, and the intergroup dependency is fused into the channels in the groups, causing mutual interference of information. The GI operation is a remediation method; thus, the loss of intergroup dependency is inevitable. An improved method called FI dilated convolution was proposed; it separates the local dependency completely from the source. In this work, the FI dilated convolution and U-Net-basic were combined into a model called U-Net-FI.
The FI dilated convolution is based on the decomposition operation of dilated convolution (Fig. 5). During the periodic subsampling of the input feature map, the neighborhood information of each channel is obtained using depthwise convolutions [39], and the feature maps obtained from the two methods are concatenated into intermediate feature maps. These groups of intermediate feature maps all have double the number of channels of the original image. Then a shared standard convolution is applied to the intermediate feature maps to produce smaller feature maps. Finally, the feature maps are re-interlaced to produce the output feature map. The FI dilated convolution adds neighborhood information as additional channels to the intermediate feature maps, avoiding the loss of intergroup dependence from the source.
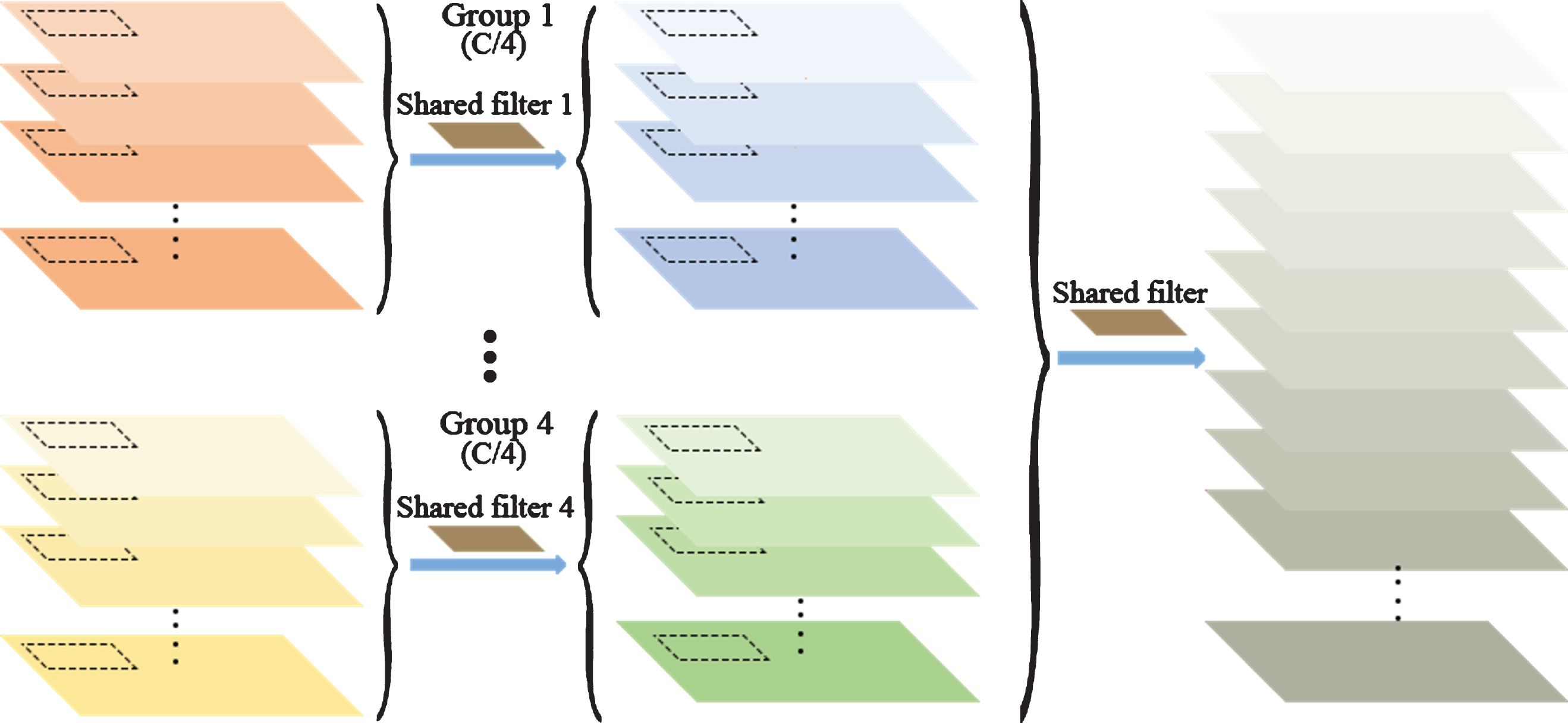
Group shared convolution.
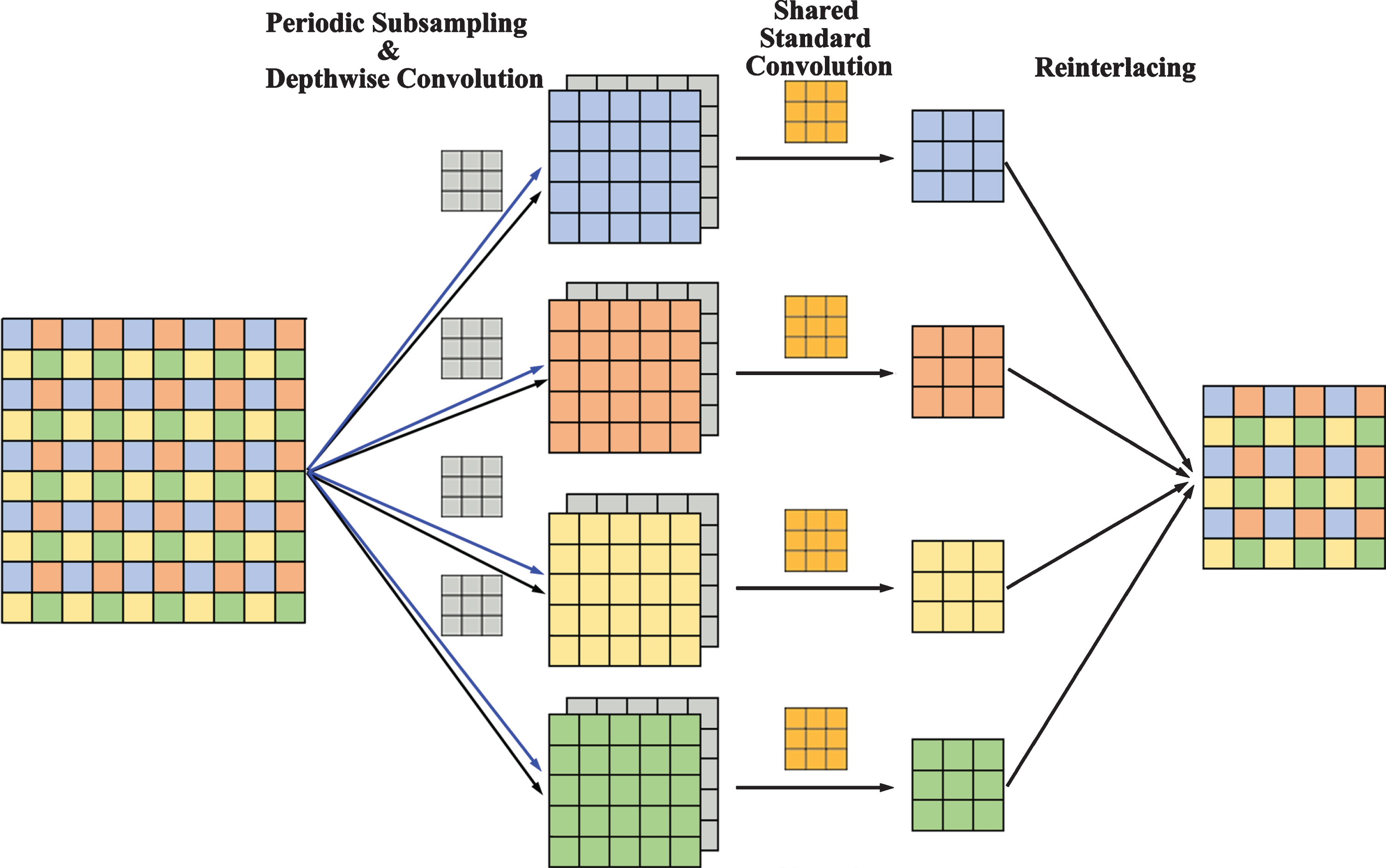
A full information dilated convolution with a kernel of 3×3, a dilation rate of r = 2, and a 2-D input feature map. The improved dilated convolution has four parts: periodic subsampling, depthwise convolution to obtain neighborhood information, shared standard convolution, and re-interlacing.
The GI operation and FI dilated convolution are very effective and can complement each other in the structure; therefore, they were combined to improve the performance. The GI operation was added before the re-interlacing of the FI operation to rebuild the dependencies among the different groups of the decomposition (Fig. 6); this new convolution operation was called the FIGI dilated convolution. As mentioned earlier, the FIGI dilated convolutions and U-Net-basic were combined into the model U-Net-FIGI.

An example of a full information dilated convolution with group interaction.
The proposed method was evaluated using two publicly available datasets. A computer (Intel i7-7700 CPU, 8 GB ram, NVIDIA GeForce GTX 1070) was used in the experiment. Python 3.6.7, tensorflow1.3.0, and pycharm 2017.2.4 were used in an Ubuntu 16.04 operating system. Without any preprocessing, chromosome segmentation was completed using the proposed end-to-end segmentation network. The segmentation network divides the overlapped chromosomes into an overlapping region, the first chromosome, the second chromosome, and the background so that the recombination of chromosomes can be easily achieved.
Performance metrics
The models were evaluated using widely used measures of overlapping chromosome segmentation. The accuracy was used as a quantitative measure to estimate output; it is calculated as follows:
The metric used to estimate each part of the segmentation result is the Jaccard similarity coefficient, which is also known as Intersection over Union (IOU); it is defined as:
Overlaps-13434 Dataset
The proposed models were trained and validated on a dataset (called Overlaps-13434) that is publicly available on Kaggle [37]. Individual chromosome images obtained from the microscope were downsampled by a factor of 2, rotated 9 times by 2π / 9, translated 5 times horizontally and vertically, and overlapped to produce samples. As a result, there were 13434 pairs of overlapping chromosomes in this data set.
The performance of all proposed models was compared using the widely used overlapping chromosome performance measures IOU, mean IOU, and accuracy (Table 1). The U-Net-basic model without any dilated convolution provided the worst performance. The reason is that the high feature variability in chromosome images requires a deep network architecture and large kernels with large receptive fields to obtain full feature representation. Although long-range information on deep blocks can be obtained through concatenated convolutions with small kernels, it requires a lot of labeled data and memory resources. Therefore, this strategy has limitations. Since large kernels with large receptive fields have been used successfully in semantic segmentation, and it is difficult to obtain labeled data of chromosomes, dilated convolutions were used in the later models.
Performance of the proposed models in stable epochs (epochs 30 to 50)
Performance of the proposed models in stable epochs (epochs 30 to 50)
Both U-Net-SS and U-Net-GI achieved better performance than U-Net-basic (Table 1). This result shows that the use of dilated convolution for the segmentation of overlapping chromosomes provides superior results, and the improvement of dilated convolution has the potential to improve the performance of the models. Therefore, the proposed improved dilated convolutions were used in the following models.
In the U-Net-GS model, the GS convolution operation was used to deal with the group dependence of the dilated convolution, and better performance than U-Net-SS and U-Net-GI was obtained (Table 1). The reason for this result is that the smoothing operation of GS convolution is gradually achieved, which can alleviate the sharp decrease in local dependent information in different channels.
U-Net-FI also achieved better performance than U-Net-SS and U-Net-GI (Table 1). The FI convolution in U-Net-FI adds neighborhood information as additional channels to the intermediate feature maps, avoiding the loss of intergroup dependence from the source.
FIGI convolution in U-Net-FIGI combines the advantages of both FI operation and GI operation; therefore, U-Net-FIGI yielded the best performance among all models (Table 1). A comparison of the performance of U-Net-basic and U-Net-FIGI in 50 epochs indicates that the use of dilated convolution provides stable performance improvement (Fig. 7).
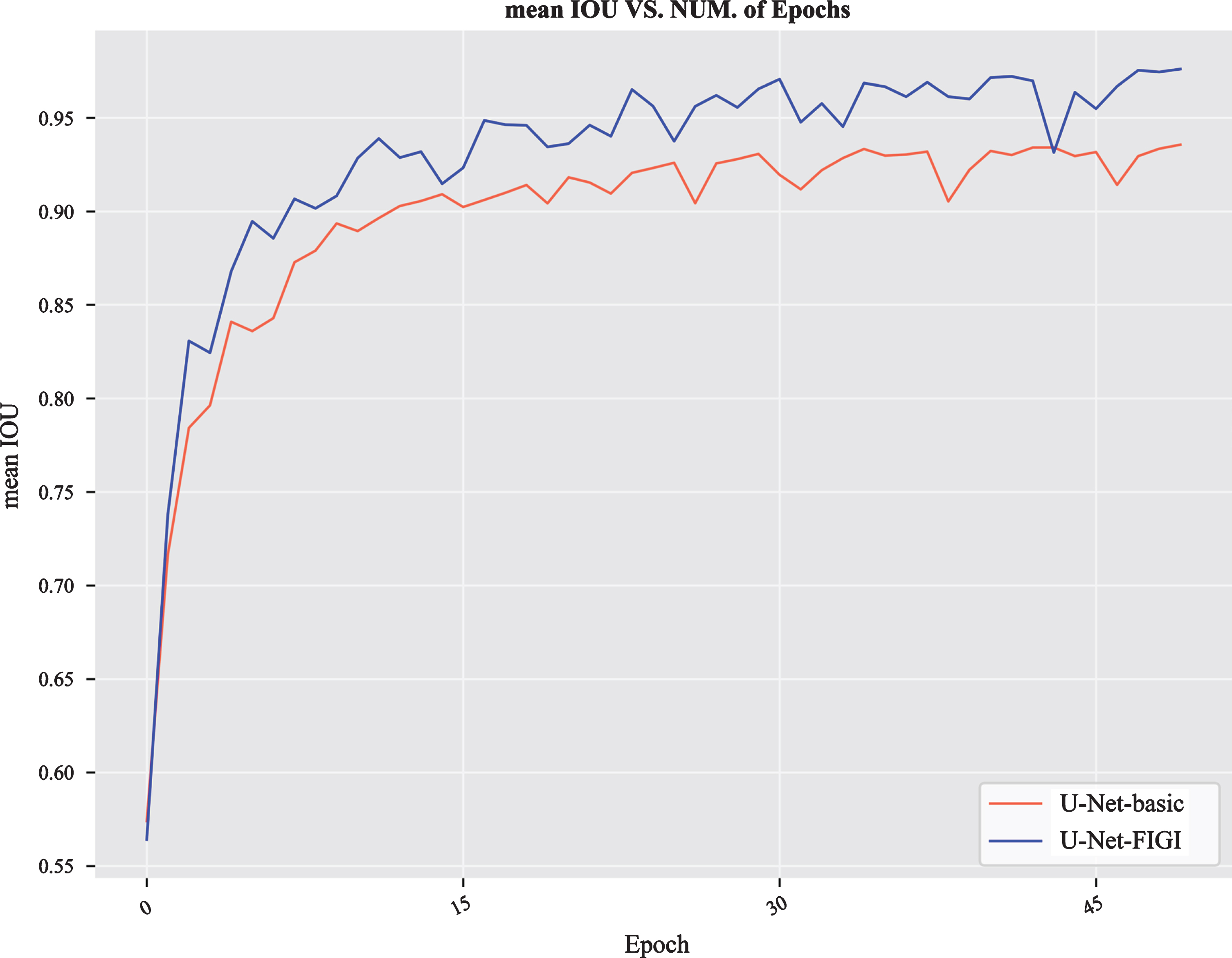
The performance of U-Net-basic and U-Net-FIGI on the Overlaps-13434 dataset for 50 epochs.
The performance of U-Net-FIGI was compared to the three latest U-Net-based methods [23, 24] using the performance measures IOU and accuracy on the Overlaps-13434 dataset to validate the performance of the proposed overlapping chromosome segmentation models based on improved dilated convolutions (Table 2). It is evident that our model provides better results for all performance measures than existing methods. In fact, all of the proposed models based on the improved dilated convolutions performed better than existing methods (Tables 1, 2). This result proves that the use of the improved dilated convolutions has significant potential to improve the segmentation accuracy of overlapping chromosomes.
Comparison of the performance measures of existing methods based on U-Net and the proposed model
Performance of the proposed models in stable epochs (epochs 30 to 50)
Experiments were also conducted on a data set called Overlaps-90667 [38], which was generated from microscope images according to the method of Ref. [45]. Individual chromosome images were downsampled by a factor of 2, rotated 18 times by 2π / 18, translated 9 times horizontally and vertically, and overlapped to produce samples. There were 90667 overlapping chromosome samples in this dataset.
All the proposed models yielded better performance on the Overlaps-90667 dataset than the Overlaps-13434 dataset (Table 3). The diversity and number of training samples of Overlaps-90667 are much higher than those of Overlaps-13434. Because the quality of the model often depends on the quality of the training data, it is natural that the data augmentation of Overlaps-90667 results in brings performance improvement. The augmentation of the chromosome data enabled the models to learn more problem-related features and prevented the models from learning too many irrelevant features, resulting in stronger generalization ability and a significant improvement in the performance of the models. The U-Net-basic model exhibited the largest performance improvement, demonstrating that data augmentation is an ideal strategy when the model is inferior.
Even in the case of data augmentation, the overlapping chromosome segmentation models based on the improved dilated convolutions still had better performance than U-Net-basic, which proved that superiority of the improved dilated convolutions. U-Net-FI performed best among all models, indicating that it is advantageous to concatenate neighborhood information into intermediate feature maps. U-Net-FIGI also performed well, and the performance difference between it and U-Net-FI was very small. U-Net-FIGI was more effective than U-Net-basic in 50 epochs (Fig. 8). The performance of these models trained using this dataset was much higher than that of existing methods (Tables 2, 3).
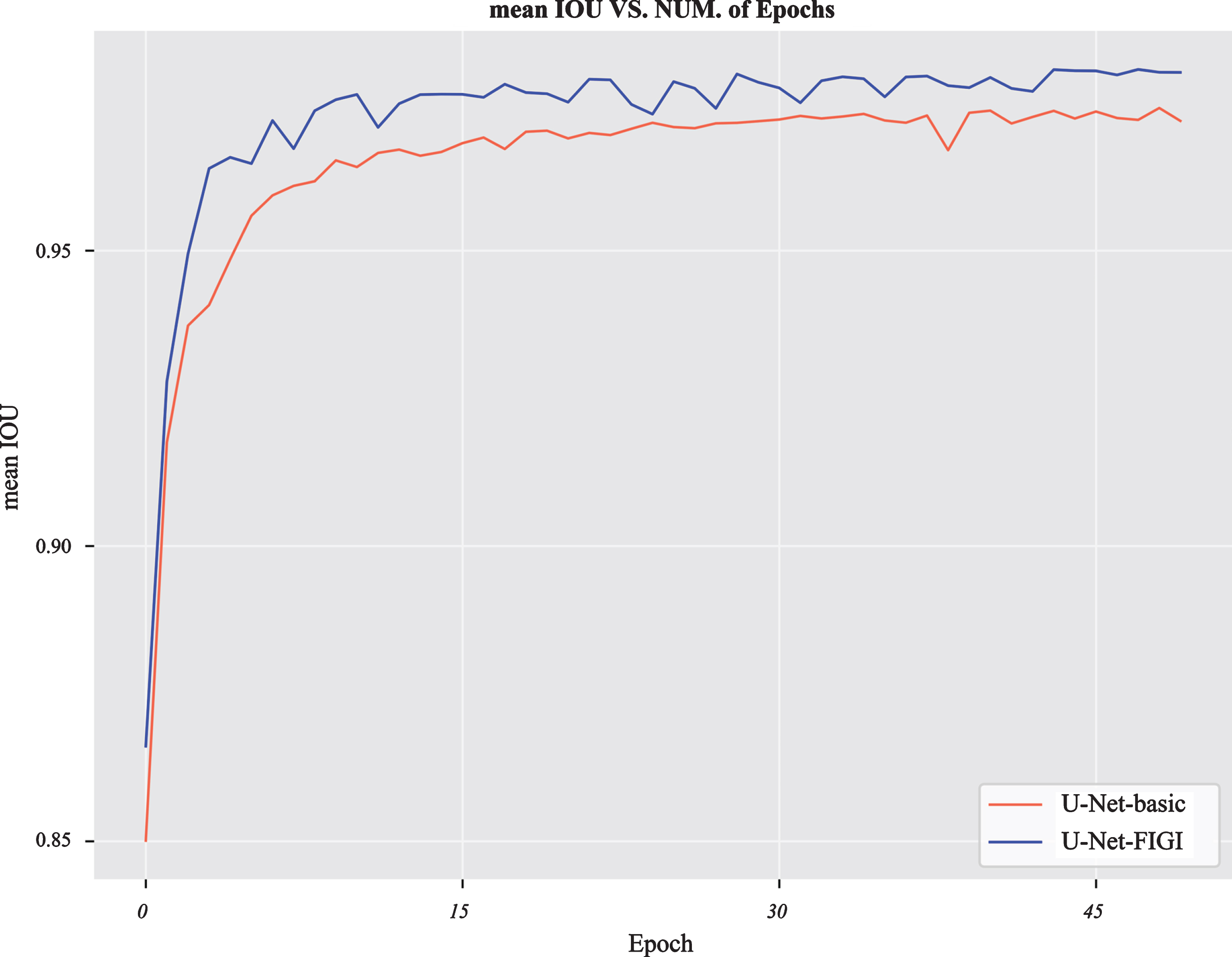
The performance of U-Net-basic and U-Net-FIGI on the Overlaps-90667 dataset for 50 epochs.
Figure 9 shows an example of the segmentation results of overlapping chromosomes by Ref. [23], U-Net-FI, and U-Net-FIGI. The first column is the background image, the second and third columns are the non-overlapping regions of the first and second chromosomes, the fourth column is the overlapping region, the fifth column is the predicted multilabel, and the last column is the ground truth. The segmentation result of Ref. [23] is in the first row. Its predicted multilabel contains impurities and is significantly different from the ground truth. This result is due to the inaccurate prediction of the first and second chromosomes, as well as the overlapping region. The second and third rows are the segmentation results of U-Net-FI and U-Net-FIGI, respectively. The multilabels look very similar to the ground truth. Figure 10 shows an example of a failed segmentation. The main reason for the segmentation failure is that the two chromosomes are similar in size and shape, and there is too much overlap and too few training samples. Increasing the number of the samples will prevent a failed segmentation.
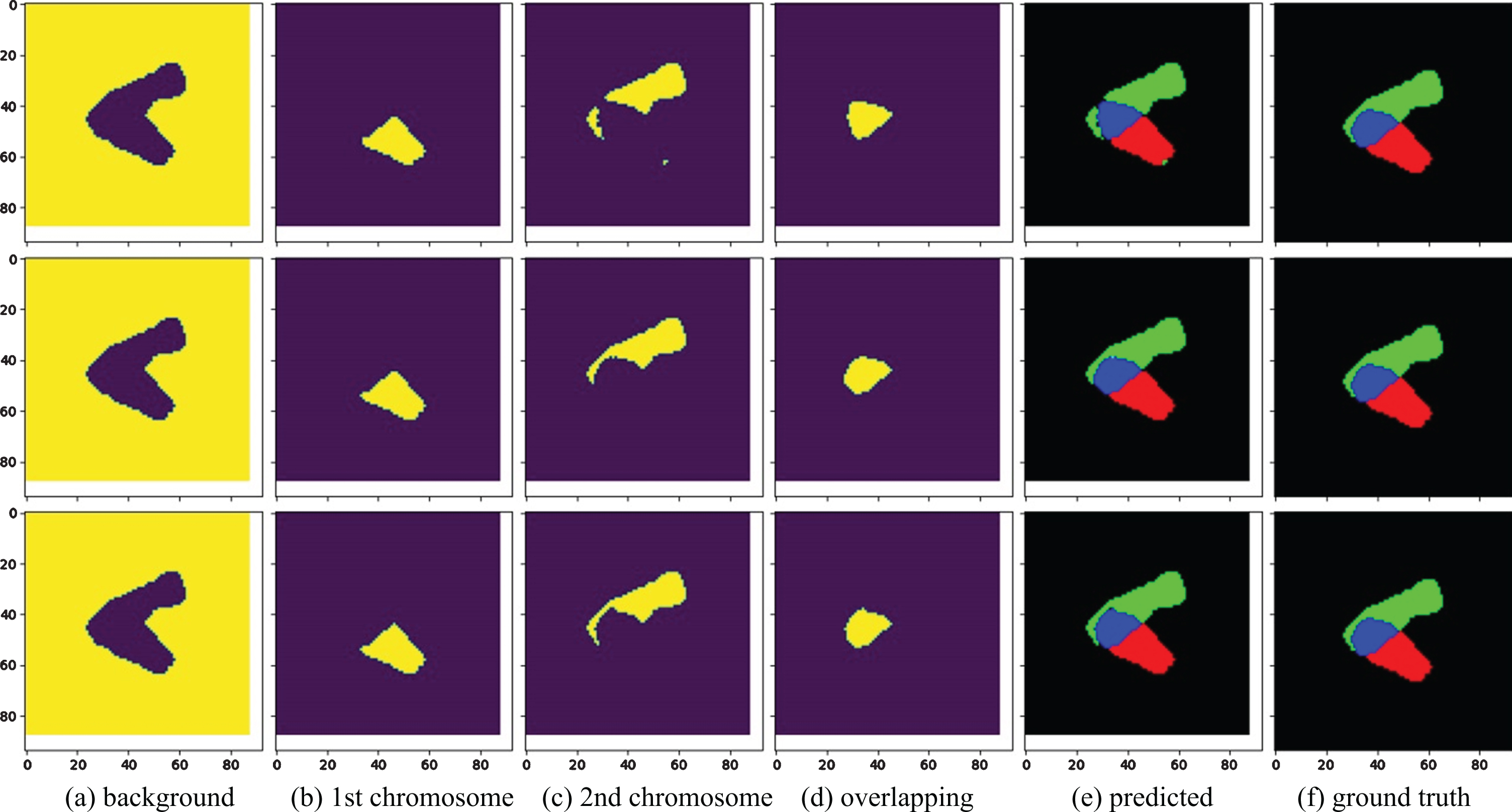
An example of segmentation results of overlapping chromosomes by Ref. [23], U-Net-FI, and U-Net-FIGI.

An example of a failed segmentation. The first row is the result obtained by Ref. [23], the second row is the result of U-Net-FI, and the third row is the result of U-Net-FIGI. Although the segmentation of this image failed, our proposed models U-Net-FI and U-Net-FIGI still performed better than the method used by Ref. [23].
U-Net-based deep CNNs were used for the segmentation of overlapping chromosomes, which is a highly complex task and remains a challenging problem. The existing SS dilated convolution and GI dilated convolution were combined with U-Net to create models for the segmentation of overlapping chromosomes. Three improved dilated convolutions were proposed, i.e., GS, FI, and FIGI dilated convolutions, which were used to create three models for the segmentation of overlapping chromosomes. The performance of the U-Net segmentation model was compared with and without the improved dilated convolution. All the proposed models with the improved dilated convolutions achieved better segmentation results than the U-net basic model. The next step is to improve the efficiency and performance of the models and apply the models to the segmentation of multiple overlapping chromosomes.
Footnotes
Acknowledgments
This work was supported by the Industry-University-Research Cooperation Project in Guangdong Province (2017B090901040) and the Key S&T Special Projects of Guangdong Province (2017B030306017, 2018B010108001).