
Abstract
Horizontal gene transference is a biological process that involves the donation of DNA or RNA from an organism to a second, unrelated organism. This process is different from the more common one, vertical transference, which is present whenever an organism or pair of organisms reproduce and transmit their genetic material to the descendants. The identification of segments of genetic material that are the result of horizontal transference is relevant to construct accurate phylogenetic trees, on one hand, and to detect possible drug-resistance mechanisms, on the other, since this movement of genetic material is the main cause behind antibiotic resistance in bacteria. Here, we describe a novel algorithm able to detect sequences of foreign origin, and thus, possible acquired via horizontal transference. The general idea of our method is that within the genome of an organism, there might be sequences that are different from the vast majority of the remaining sequences from the same organism. The former are candidate anomalies, and thus, their origin may be explained by horizontal transference. This approach is equivalent to a particular instance of the authorship attribution problem, that in which from a set of texts or paragraphs, almost all of them were written by the same author, whereas a minority has a different authorship. The constraint is that the author of each text is not known, so the algorithm has to attribute the authorship of each one of the texts. The texts detected to be written by a different author are the equivalent of the sequences of foreign origin for the case of genetic material. We describe here a novel method to detect anomalous sequences, based on interpretable embeddings derived from a common attention mechanism in humans, that of identifying novel tokens within a given sequence. Our proposal achieves novel and consistent results over the genome of a well known organism.
Introduction
The genome of an organism consists of its nucleotide content [1]. Depending on the nature of the organism, its genomic material can be either DNA or RNA, that is, deoxyribonucleic or ribonucleic acids. In either case the, nucleotides that conform the genome are four possible bases or nucleotides: Adenine (A), Guanine (G), Cytosine (C) and, for the case of DNA Timine (T) or Uracil (U) for RNA. The genomic sequences are thus represented by a usually long sequence of nucleotides. For instance, the human genome consists of 3 × 109 base pairs (bp) [2], and the genome of the bacteria Mycobacterium tuberculosis, which will be of high relevance in this contribution, consists of over four million bp [3].
The genetic material of an organism is almost always directly inherited from its ancestors. This process is known as vertical transference, since it goes from an organism to its descendants. In some organisms, there is a second path to acquire genetic material. Some organisms may transmit part of their genetic material to other, unrelated organisms after the receiver has matured [4]. This process is called horizontal gene transference (HGT), and it is also referred to as lateral (as opposed to vertical) transference. A gene is a sequence of nucleotides that code the amino acids that are needed to form a protein. Proteins are essential for life as we know it, since they provide support to the cells, means of for signal processing, protection against foreign organisms, and many more functions [5]. In this contribution we will stick to nucleotide sequences and thus no further reference to proteins will be made.
The detection of genetic material that was acquired by HGT is of high relevance for at least two reasons. The first one is related to Natural History. Biologists are interested in finding lateral branches in the tree of life. This phenomena sheds light into the complex and convoluted evolutionary process in organisms [6], and it also helps to explain the appearance of new traits in organisms. The second reason behind the interest in detecting HGT material is of medical nature. Some bacteria are known to present resistance against antibiotics or some other drugs after acquiring some genes that were transferred to them by organisms that were already drug-resistant [7]. The donor organisms may be unrelated to the recipient organism, and the HGT process can occur via several mechanisms, which can be summarized as a direct transfer from donor to receiver, or via a modification of the genetic material [8]. Once an organism, say, a bacteria, acquires a drug resistance gene, it can be further transmitted to other bacteria by HGT. This latter consequence of HGT may be a threat to public health, as it affects the policies to treat bacterial infections.
The identification of sequences or genes acquired by HGT is of high relevance in the Biomedical sciences. The computational problem of the identification of HGT is in itself of high interest, but it has to face several constraints. First, there is no comprehensive database that covers all genes and organisms. One of the most complete ones is that of [8, 9], and yet it does not allow the use of, for example, classification tools to tell apart genes acquired from lateral or horizontal transference. Second, since the number of genes that are known to be horizontally acquired in an organism is rather small, there is a lack of samples to accurately train an algorithm,
In this contribution we approach the HGT problem following a perspective supported by two areas in Artificial Intelligence. The first one is derived from Natural Language Processing (NLP) and the second one comes from unsupervised learning, in particular, from anomaly detection. The general idea behind our HGT detection method can be depicted in a NLP problem as follows. Consider a set of texts almost all written by the same author. If we are told that one or a small proportion of the texts were written by a different author, but we are not told which, we can apply tools related to the authorship attribution problem [28, 29]. A possible approach would be to detect some attributes that are common in the majority of the texts, but are not to be found in a few of them [30]. The former set would constitute the group of the texts written by the same author, whereas the latter would constitute the set of texts with a different authorship.
In the HGT context, we do not have authors, but organisms. We do not have texts but sequences and genetic material. If we abstract the contexts of these two fields, it quickly becomes clear that the problem of HGT is rather similar to the authorship attribution problem just depicted. Now, since our method identifies some features that are common in a large subset of the samples, being texts or genomic sequences, but are not present in a small subsample, and we are not told which are which, the problem falls into the anomaly detection field, the second leg of our approach.
An anomaly is an instance that is different from the rest of the observations within a dataset [31]. Although we will elaborate in this regard in the following sections, for the time being, suffice it to say that within a dataset, an unsupervised learning algorithm can rank the level of anomaly of each vector or observation. In the HGT problem, the higher the anomaly level of a sequence from an organism, the more likely that sequence is of foreign origin, that is, there is more evidence that it was acquired by HGT.
The HGT detection algorithm we present in this contribution is useful in at least three fronts. In the first one, it frames the HGT problem into an anomaly detection perspective. Thus, several existing tools can be applied. Here, we focus our efforts in only one anomaly detection algorithm, but many extensions are possible. Second, our method is explainable. The descriptions achieved by our method provides an explainable embedding, which may help biologists and geneticists to explain why some genomic regions were identified as anomalies, and thus, probably obtained by HGT.
Our method is strongly based on the positional information of tokens within a sequence. The positional information simply refers to the order of appearance of the tokens within a sequence. In the HGT context, a token, as described in the next sections, is a possibly short sequence of nucleotides. Based on the positional structure, the concept of distance between tokens can be induced. The distance between a token in position i and a token in position j is simply |j - i|. Our working hypothesis is that by carefully selecting some characterization of the positional information, it is plausible to map sequences to an attribute space from which sequences within a genome can be identified as acquired by HGT or by vertical transference. In parallel, such embedding may allow specialists to look into genomes from a different perspective, and perhaps more unveiling perspective.
The rest of the contribution continues as follows. In Section 2 we briefly describe the state-of-the-art of HGT algorithms. In Section 3 we describe our proposal to deal with the HGT problem, and we further proceed to present some results in Section 4. Finally, we offer some discussion and conclusions in Section 5.
State-of-the-art
There are several families of HGT detection algorithms. In general, the assumptions behind those algorithms can be summarized into the following: 1. Genes acquired by HGT present an evolutionary history different from the rest of the genome. 2. HGT events can be detected since they leave a mark in the genome. Based on these two assumptions, several alternatives have been proposed [11].
Common approaches to detect HGT compare a certain gene in an organism of a certain phylum or species to genes in other, unrelated organisms [10]. If there is a match, generally computed by the dynamic programming Needleman-Wunsch algorithm [12], then both organisms might either descend from a common ancestor, or one of the organisms donated the gene to the other. In general, it is not clear which is the donor and which the receiver organisms, so further investigations in the phylogeny are in order.
In [14], authors sequences are inspected in order to determine weak signals of changes in synteny, which is the conservation of gene order across species along the evolutionary course. That is, by detecting the position of a gene within a genome, authors identify those organisms in which a gene occupies a position different from the position that same gene occupies in a different organism. In [15], authors define a robust HGT identification framework based on graph theory, and by recreating the most likely paths within a graph, identify the genes that are more likely to have been acquired by HGT.
In [13], authors analyze changes in the frequency of use of codons usage of genes. Codon usage is the relative frequency of appearance of each triplet or token of three nucleotides. Genes with a different codon usage that the expected frequencies within a genome are likely to be the result of an HGT event.
There are many more HGT detection algorithms, and the vast majority of them make assumptions about the relative frequencies of tokens or short sequences. In our approach, the frequency of appearance of the those tokens is not relevant. What is relevant is the distance, or number of tokens that separate a given token from its previous appearance. Equally relevant for our approach is the number of tokens separating a certain token from the last novel one. This will be detailed in the next section.
Our algorithm
Let
Let U be the set of all sequences U = W0, …, WL-1. U is formed by two mutually exclusive sets, A and S. The set A is formed by all sequences that were obtained by horizontal transference, whereas the set S contains the sequences acquired by vertical transference. Let X be the list of attributes extracted from the sequences in S, such that, there exist a function

The problem is two-fold, since neither X nor
An anomaly is an instance, observation or vector that does not resemble the rest of the observations within the dataset under study [32]. An anomaly detection algorithm (AD) aims to identify some attributes that are common in the majority of the elements under study, and are not present in a, generally small, subset of elements in the same dataset [33]. The problem can be stated as identifying a list of attributes, and a decision procedure, to tell apart expected, usual or normal observations, from the anomalous or unexpected instances. AD is in general an unsupervised task, since vectors are not assigned an external label or class. If that were the case, the AD problem would be approached following a supervised learning approach.
AD algorithms in general rank vectors accordingly to their anomaly factor. The anomaly factor can be computed in several ways [34]. Once all vectors are ranked, a decision about the threshold of expected or usual vectors is taken, frequently on probability assumptions.
Lets return to our interest in HGT detection. A sequence
Let K0 be the first lexicographical token. For instance if k = 3, then K0 = AAA, K1 = AAC, . . . , K63 = TTT. A sequence W
i
can be characterized in terms of some properties of its constituents, that is, its kmers. A succinct representation of a sequence W can be obtained by referring to its tokens. For example, let W
i
= AATCGTTTA be a sequence in
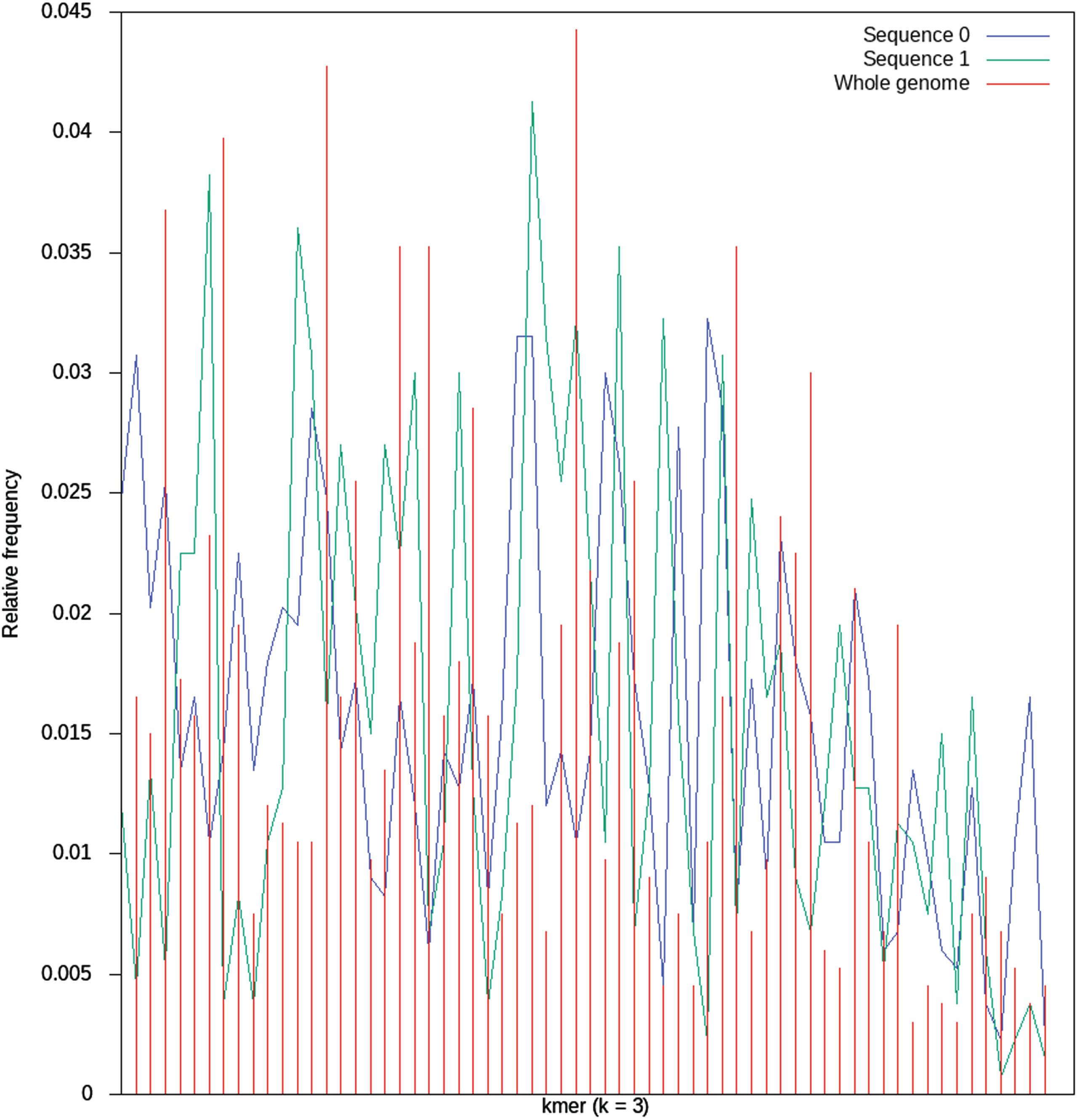
Codon usage of Mycobacterium tuberculosis, for its whole genome and for some sequences of length 3,500bp. The codon usage of a sequence is the relative frequency of appearance of each of its kmers. In the figure, it is shown this information for k = 3, that is, for the 64 possible kmers (43).
Remember that our task is to identify the sets A and S, that is, the set of genetic material of possibly external origin, and the set of common or own sequences. Some approaches have proposed the codon usage of sequences in order to approximate A and S [13]. If the comparison of two histograms representing the codon usage of two genes is similar, then a HGT phenomena might had occured between the two organisms [16].
Although codon usage has shown some relevant results, there are other alternatives to characterize sequences. Our approach goes in a different direction, based on aspects of cognitive computing and NLP. In order to depict its general ideas, imagine you are inspecting a sequence of tokens. It is a common and relatively easy task to remember when the token you are looking at appeared for the last time within that sequence. It is also a relatively simple cognitive task to identify the whether the current observed token is a novel one or not. By novel we refer to a token that appears for the first time within the sequence. It is from these two observations that we construct our proposal to create X, the set of attributes that will help us to tell apart the sets A and S, that is, sequences acquired by HGT, and sequences inherited by vertical transference, respectively. From this characterization, we base our definitions in a recent article [17], where the problem of authorship attribution was tackled with the embeddings defined in the next paragraphs.
Let σ (K i , K j ) be the distance between kmers at positions i and j in W, respectively. Note that the sub-index refers to a specific kmer, whereas the super-index refers to the kmer at the indicated position. σ (K i , K j ) is defined as the number of kmers that separates positions i and j, that is |i - j|. From our description in the previous paragraph, we are interested in the distance between the kmer at position i, K i and the previous appearance of that same kmer, that is K K i . We will denote this distance as δ (K i ). If it is the first appearance of kmer K K i , then δ (K i ) =0. In order to compare sequences of different size, δ (K i ) is escalated to the range [0, 1] by dividing it by the length of the sequence, |W|.
The algorithm analyzes W and maintains a record of the position in which each kmer appears. A kmer is declared novel if it appears for the first time in W. We will identify the novel token as η. At first, η = K K 0 , that is, the novel token is the first obseved kmer. If the second kmer is not the same as the first one, η = K K 1 . The position of η is omitted for clarity, but it is implicitly maintained for comparisons. The positions of each kmer goes from 0 to 9. Table 1 shows as an example the analysis of the sequence K3K2K9K3K2K17K38K52K41K41.
An example of the characterization of the sequence W = K3K2K9K3K2K17K38K52K41K41. It is shown the last novel kmer η for each kmer in position i, K i , the distance to the previous appearance of the same token K K i (δ (K i )), and the distance to the last novel kmer, σ (K i , η). The position within W of η is not shown for clarity, but it is used to compute σ (K i , η)

Each kmer K in W is characterized by two attributes. The first attribute is the expected value of the distance between K and its previous appearance, and the second feature is the expected value of the distance from K to the last novel token. More formally, token K is characterized as:
Let ∈
K
be the number of appearances of kmer K in W. Then, since δ (K, p) refers to the distance to the last appearance of kmer K, when it appeared at position p, we have: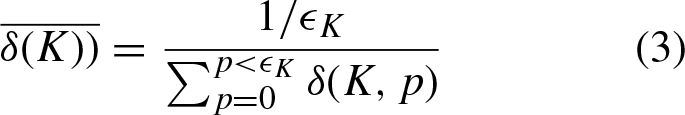
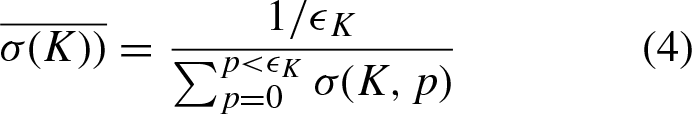
That is, we simply compute the average distances from a token K to its previous appearances, on one hand, and the distance from that token to the last novel kmer, over all the appearances of K, on the other. Thus, each kmer K is described by two attributes, and thus, can be represented in a two-dimensional space. Figure 2 shows the embedding of all 64 kmers for k = 3 for two sequences W0 and W2 from the genome of Mycobacterium tuberculosis.

The embedding formed by the expected distance from a kmer or token to its previous appearance within a sequence (x-axis) and the expected distance from the kmer to the last novel kmer. It is shown the result for the genome
So far, we have characterized each token K in a sequence W
a
as X
K
. Now, we have to create a final embedding or description for W
a
. To do this, we simply join the description of all 4
k
possible kmers into one single vector, and we call it 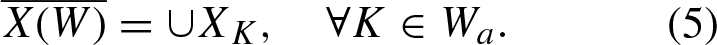
Finally, we apply an anomaly detection algorithm over the corresponding descriptions of all sequences in
In this contribution, we applied Isolation Forests (IF) [18] as the algorithm to detect anomalies. The basic idea behind IF is that the more difficult is to randomly isolate a vector from the remaining ones within the same dataset, the more less anomalous is. On the other hand, if a vector is easily isolated from the remaining observations, then that vector has a higher anomaly level.
A vector v is to be isolated by making cuts in the N-dimensional attribute space. The hyperplane of dimension N - 1 in which the projection has to be made is selected at random, and then from the range of possible values, a threshold is randomly selected. Figure 3 depicts the general idea behind IF. Since the process is stochastic, several Monte Carlo experiments have to be made in order to approximate the true difficulty of isolation for each vector. The anomaly level assigned to a vector v is a measure of the expected size of the tree needed to isolate v from the rest of the vectors in the attribute space.

Isolation Forests (IF) for anomaly detection. The number of cuts or decisions that have to be made in order to create an area in which only one vector is located is a measure of the anomaly factor associated to it. On top, it is shown a random series of cuts to isolate the orange vector, and the associated tree. The bottom figure depicts a series of random cuts to isolate the blue vector. The former requires less cuts than the latter, and thus, its anomaly factor is higher. The less random cuts are needed to isolate a vector, the higher its anomaly factor or level.
In the case of HGT, we made the assumption that sequences within a genome that are identified as anomalies, are candidates of being acquired by horizontal transference. So far, we have described the set of attributes X that characterize sequences within a genome. If we inspect this new set of attributes, we can detect those sequences whose description differs from that of the majority of sequences. The simplest scenario, to which we stick for simplicity, is to sort sequences based on the anomaly criteria. The most anomalous sequences are the anomaly candidates, and they can be inspected in both, the literature, and in some existing databases of HGT evidence.
In this way, we have completed our journey to elucidate an algorithm able to detect HGT, based on anomaly detection of NLP-inspired attributes derived from genomic material. In summary, our approach does the following. Given a genome of interest,
Mycobacterium tuberculosis is the causal agent of Tuberculosis (TB), an infectious disease that causes over two million deaths worldwide [19]. The impact in humans suffering from TB is acute, affecting mainly the lungs and the respiratory tract, among other organs and tissues. The quality of life of individuals suffering from TB that are not under medical surveillance can be reduced [20]. It is estimated that, because of drug-resistance in Mycobacterium tuberculosis, the number of yearly cases may witness an abrupt increase within the next few decades. Thus, it is of high relevance to detect HGT in this, and other related bacteria.
The genome of Mycobacterium tuberculosis consists 4,417,942 bp. There are at least two reference genomes, that is, genomes that have been assembled and, although both are rather similar, we will focus our analysis in the H37rv strain. This strain contains around 3,900 genes, with an average size of just over 3000bp [21].
We describe some aspects of Mycobacterium tuberculosis because this is the organism we selected to test HorGenTIAD. It is kown that some genes in this organism are of foreign origin, as stated in [23]. We conducted several tests for the size of the sequences to split

The anomaly factor, obtained by IF, of each of the 1019 sequences of size 3,500 base pairs (nucleotides) of the Mycobacterium tuberculosis genome. The embedding space
We would like to remind the reader that the genome
The red markers indicate genes that are plausible, accordingly to the literature (see Table 2), to be the result of an HGT event. In green, it is shown putative or candidate genes from which no evidence was found of their foreign (HGT) origin. It is important to note that this sequences, in general, do not have a gene name, although might have sequence identifiers. The fact that they do not have a name indicates that further characterizations are in order to fully understand their function and whether they code a protein or not. The blue marker indicates a (proper) gene that is not of HGT origin.
Now, Fig. 5 shows the result of applying IF over a different embedding, that of codon usage, as indicated by [16]. It can be seen that the sequences with the highest anomaly factor contain in general putative genes, whose origin cannot be confirmed. Besides, two genes of lateral origin were detected as anomalies. This offers insight into the difficulties of relying solely on codon usage information for HGT detection.

The anomaly factor, obtained by IF, of each of the 1019 sequences of size 3,500 base pairs (nucleotides) of the Mycobacterium tuberculosis genome. The embedding space is the codon usage (codon frequency) of each kmer. In red it is shown the true positives, accordingly to the literature, and in blue the false positive. In this context a true positive is a sequence that contains material of foreign origin, that is acquired from a HGT event. A false positive is a sequence identified as HGT, when there is no evidence to support it. The green elements indicate sequences that were identified as anomalies (HGT) but there is not enough characterization of the (putative) genes as to clarify it. For this case, no genes known to be of foreign origin were correctly identified, and the number of false positives is higher that for the previous case.
Horizontal gene transference in Mycobacterium tuberculosis has been difficult to detect for a number of reasons [36]. First, no recent HGT events have been detected [35], and second, there is no consensus from the existing methodologies [37]. In order to compare the results from HorGenTIAD to those achieved by other methods, we compared the genes that were detected as acquired by HGT by our method to those detected as such in the database described in [38] and by the codon usage method [16]. Note that there are no ground truths for the case of HGT at least not for Mycobacterium tuberculosis, and thus, metrics such as recall and precision are not relevant.
Figure 6 shows the comparison of HorGenTIAD and the two common approaches mentioned above. It is displayed the number of overlapping genes for the three approaches, and their combinations. It is observed that HorGenTIAD detected the lowest number of HGT events, with 146 genes, whereas the number of HGT events detected by the remaining two methods (Friedman and Garcia) are 195 and 196, respectively. However, the number of common genes in these two methods is only of 33. The number of HGT events detected as such by the three approaches is rather small, since only thirteen genes were identified as acquired by HGT.
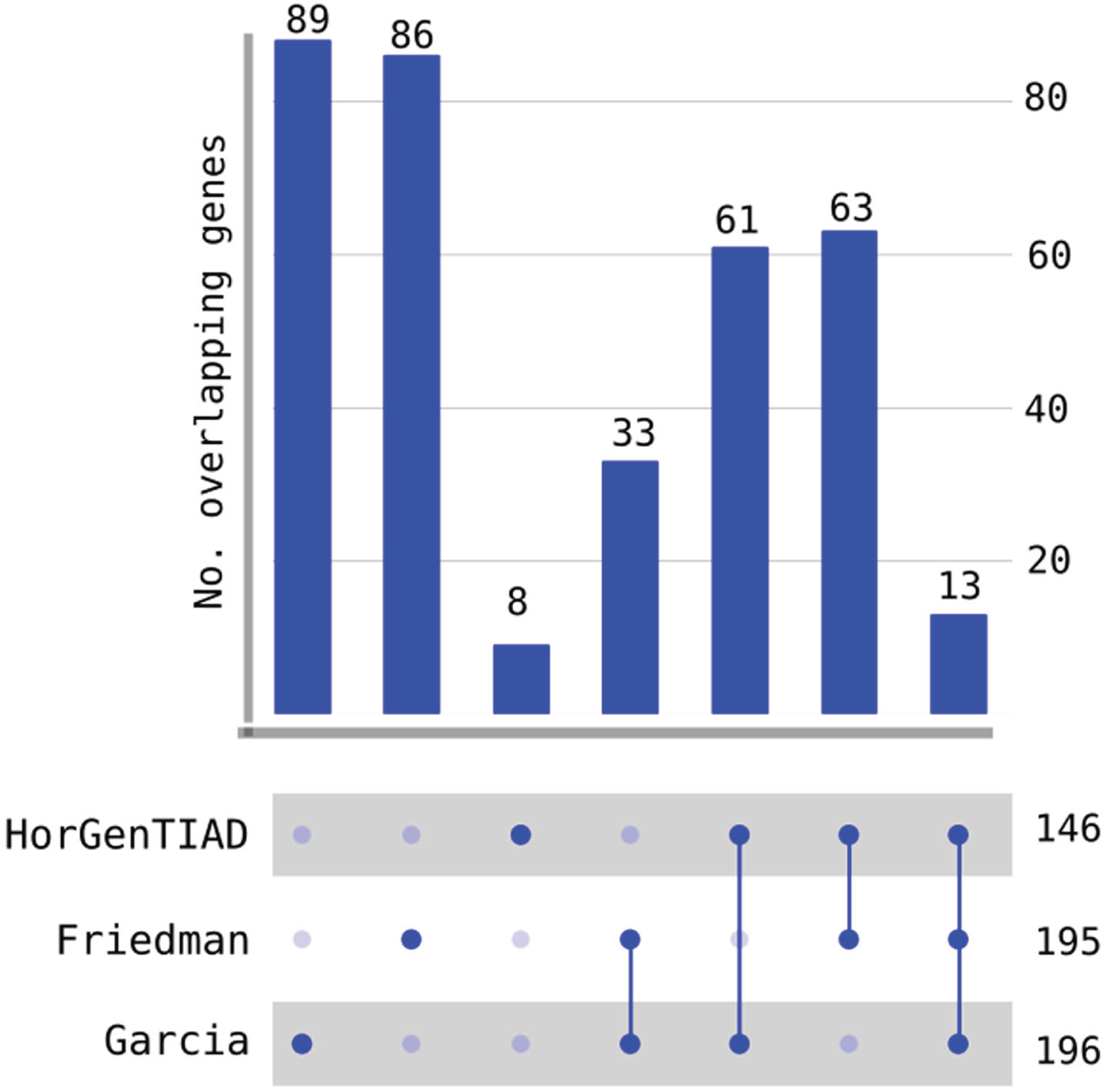
Comparison of HorGenTIAD, Friedman [16] and Garcia [38] methods in the detection of HGT in Mycobacterium tuberculosis. HorGenTIAD detected 8 genes that were not detected by the remaining two methods, whereas, in total, this method detected 146 genes. The Garcia and Friedman methods detected a larger number of genes (196 and 195, respectively), although the overlap of the two methods is low (33 when HorGenTIAD was not considered).
A relevant case is that of the eight HGT events detected only by HorGenTIAD. Those 8 genes are precisely those shown in Fig. 4, whereas the labelled genes in Fig. 5 are a subset of the 86 genes detected by the Friedman method.
Since there are no comprehensive databases to systematically validate the identified genes as acquired by HGT, we consulted the literature to validate our findings. Table 2 offers some details of five of the eight genes detected by HorGenTIAD. The table offers a summary of the genes detected as acquired by HGT from the
Genes contained in sequences identified as anomalies, that is, as containing HGT material. The embedding

Horizontal gene transference is a process in which genetic material is transferred from a donor to a receiver. The latter is not, in general a descendant from the former. Events of horizontal gene transference are relevant since they explain in more detail the evolution of life, in one hand, and because those events are a strong mechanism behind drug-resistance in bacteria.
The identification of horizontal gene transference events is an open problem, since in general, there is no external label or class assigned to all possible genes or sequences. That class, that is, the label of whether a sequence or gene is the result of an horizontal gene transference event or not, is lacking in the majority of the cases. Thus, Biologists, Computer Scientists, Mathematicians and Bioinformaticians have tackled the problem from several perspectives.
In this contribution, we have boarded the problem of horizontal gene transference detection based on elements from natural language processing and from unsupervised learning, in particular, from anomaly detection algorithms. The natural language processing elements that inspired our method are related to the authorship attribution problem. In our approach, however, we do not have authors but organisms, and we do not have texts to be attributed but sequenced to be identified as obtained by an horizontal gene transference event or acquired by the more common path of vertical inheritance. In that regard, our approach is equivalent to the authorship attribution problem in.
The genome under analysis is split into several non overlapping sequences. Then each sequence is further characterized in terms of some properties of their constituent tokens, or, as they are usually called in the molecular biology field, kmers. Each kmer is described in terms of the number of tokens separating it from the last occurrence of the same token within the current window, and also described in terms of the number of tokens separating it from the last novel token. This approach is derived from a recent embedding within the authorship attribution problem [17]. Once sequences of the studied organism are embedded in this new space, an anomaly detection algorithm is applied to all sequences. An anomaly detection algorithm aims to identify some elements within a collection that are not similar to the vast majority of the elements under study. Anomaly detection is an instance of unsupervised learning, which provides the second path of support to our method. Those sequences with the highest anomaly level, based on a specific anomaly dtection algorithm called Isolation Forests, are candidates of being of foreign origin, that is, the assumption is that they were acquired by an HGT event.
We tested our method over the genome of the bacteria Mycobacterium tuberculosis. Our method, HorGenTIAD, was able to identify sequences that contain genes from a plausible HGT origin, accordingly to the literature. We compared our method with one of the existing approaches, that f codon usage, and, in our opinion, the results are promising.
We have provided in this work enough evidence to support our hypothesis, which, to remind the reader, can be enunciated as follows. The number of tokens or kmers separating consecutive appearances of a certain kmer, together with the number of kmers separating a kmer from the last novel kmer, form the basis to generate an embedding from which we can elucidate whether a certain sequence contains material of foreign or HGT origin. Our approach is based purely in the nucleotide alphabet, and thus, no transcriptomic (protein) information is invoked, which gives our method.
Even though our results are encouraging, HorGenTIAD has to be extensively and systematically evaluated in larger datasets. The construction of HGT databases is an ongoing task. We can conclude that our method offers a novel way to inspect genomic sequences. The identification of attributes, derived from some properties of the kmers in a sequence, can help biologists to understand from a different perspective those sequences. Finally, the framing of the horizontal gene transference problem as an anomaly detection task is also, in our opinion, a step further in order to bring more computational perspectives to complex phenomena, as is the case in Biology.
Acknowledgments
Authors would like to thank DGAPA PAPIIT for finantial support under projects TA101323 and TA100721.
Availability
Software is available at https://anomalocarisproject.github.io/