
Abstract
Parkinson’s disease (PD) is in some cases predisposed-or-caused by genetic variants, contributing to the expression of different phenotypes. Regardless of etiology, as the disease progresses, motor fluctuations and/or levodopa-induced dyskinesias limit the benefit of pharmacotherapy. Device-aided therapies are good alternatives in advanced disease, including deep brain stimulation (DBS), levodopa-carbidopa intestinal gel, and continuous subcutaneous infusion of apomorphine. Candidate selection and timing are critical for the success of such therapies. Genetic screening in DBS cohorts has shown a higher proportion of mutation carriers than in general cohorts, suggesting that genetic factors may influence candidacy for advanced therapies. The response of monogenic PD to device therapies is not well established, and the contribution of genetic information to decision-making is still a matter of debate. The limited evidence regarding gene-dependent response to device-aided therapies is reviewed here. An accurate understanding of the adequacy and responses of different mutation carriers to device-aided therapies requires the development of specific studies with long-term monitoring.
Keywords
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative condition after Alzheimer’s disease and is the fastest-growing neurodegenerative disorder, with a projected prevalence of 12 million by 2040 [1]. The incidence ranges from 5 to 25 annual cases per 100,000, with the mean age of onset in the seventh decade [2].
There is still a knowledge gap in our understanding of the molecular basis for neurodegeneration in PD. Several environmental and genetic risk factors have been identified, including rare monogenic disorders [3].
A broad genotype-phenotype correlation can be recognized for certain variants [4]. Besides, monogenic PD may benefit from gene-specific treatment strategies (i.e., LRRK2 and GBA) [5]. However, it remains to be seen whether this applies to device-aided therapies such as deep brain stimulation (DBS), levodopa-carbidopa intestinal gel infusion (LCIG), or continuous subcutaneous apomorphine infusion (CSAI) [6, 7]. These therapies are potential options for individuals with PD with complications such as refractory “wearing-off” and levodopa-induced dyskinesias [8].
Surprisingly, genetic screening has demonstrated an overrepresentation of specific genetic variants in DBS cohorts (up to 29%) compared to the overall PD population (estimated at 5–10%). GBA, LRRK2, and PRKN variants are the most frequent [9].
This association raises whether individuals with certain genetic variants represent better candidates for specific device-aided therapies and whether these genetic factors affect the response to such therapies. We suggest reading a previous publication addressing the phenotype-genotype relationship in decision-making on device-aided therapies [10]. The latter issue, how genetic factors affect treatment response, is addressed below.
DEVICE-AIDED THERAPIES
The success of device-aided therapies depends on selecting the suitable device for the right patient. Key eligibility features include: (I) ≥1 hour of troublesome dyskinesia per day, or≥2 hours “off” symptoms per day and need to take levodopa≥5-times per day; (II) no more than mild dementia and absence of troublesome hallucinations; and (III) significant difficulty with activities of daily living. Patients demonstrating good levodopa response who are emotionally stable, physically healthy, cognitively intact, and younger (preferably < 70 years of age) are ideal candidates for CSAI, LCIG, or DBS [11]. While these therapies seem to provide a similar improvement in reducing “off” time by around 40–60%, their effects on dyskinesia and non-motor symptoms are heterogeneous, and their side effects and complications can be quite different [12]. Table 1 summarizes their main indications, advantages, disadvantages, and contraindications.
Comparison between different device-aided therapies
DBS, deep brain stimulation; LCIG, Levodopa-carbidopa intestinal gel; CASI, continual apomorphine subcutaneous infusion; iPD, idiopathic Parkinson’s disease; NMS, non motor symptoms; PEJ, percutaneous endoscopic Jejunostomy; AE, adverse effects; ICD, impulse control disorders; LEDD, Levodopa equivalent daily dose.
In terms of selecting which device therapy is most appropriate, DBS is favored with younger patients and minimal non-levodopa-responsive motor symptoms (except for tremor) [13, 14]. DBS may be contraindicated if there is dementia, hallucinations, uncontrolled depression, marked postural and gait problems, severe brain atrophy, or suspected atypical parkinsonism.
LCIG can still be considered for patients with mild-to-moderate dementia or age > 70 years, even with severe depression [15]. However, patients with dopamine dysregulation, punding, or pre-existent peripheral neuropathies may be less favorable candidates.
CSAI can be considered if there are mild hallucinations or moderate cognitive impairment. Moreover, it might improve neuropsychological performance [16, 17]. In addition, this device may ameliorate depression, apathy, “off” pain, and slowness of thinking [18]. However, it seems less favorable in patients with impulse control disorders (ICD), marked psychosis, daytime somnolence, and troublesome orthostatic hypotension [19].
We caution against dogmatism since there is also favorable evidence for device therapies in some of these reported contraindications. For example, there are cases where LCIG and CSAI improve ICD, and where LCIG decreases dopamine dysregulation syndrome [20–22]
A relevant factor in decision-making for device-aided therapies is the long-term outcome expectancy. Chronic STN-DBS can cause dysarthric speech, problems in verbal fluency, worsening freezing of gait, and axial symptoms. These are important determinants of quality of life [23–25]. Severe pre-operative gait difficulties might predict limited long-term DBS benefits [23, 24]. Moreover, motor outcomes one year after bilateral STN-DBS are inversely correlated with the rate of progression of motor symptoms [26].
The leading causes of discontinuation of LCIG therapy in long-term follow-up include worsening cognition, dyskinesias, chronic polyneuropathy, weight loss, and hallucinations. Eventually, LCIG may become ineffective [27, 28].
Like LCIG, long-term CSAI may worsen cognition, dyskinesias, postural instability, and hallucinations. CSAI also causes sedation and orthostatic hypotension. These factors may obscure the long-term benefits in some patients [29–32]. Furthermore, a decrease in therapeutic effect may become an important reason for discontinuation within the first four years [33].
In conclusion, it is essential to recognize patient heterogeneity and, if possible, identify biomarkers of short and long-term outcomes. Understanding phenotype-genotype relationships and how variants predict the risk of significant disease milestones [34] may affect the timing, appropriateness, expected outcome, and expectations for device-aided therapies [10].
CLINICAL FEATURES OF CAUSAL MUTATIONS AND GENETIC RISK FACTORS FOR PARKINSONISM
Most PD cases are sporadic, associated with genetic, epigenetic, and environmental risk factors [35]. The most frequent genetic risk factors for sporadic PD are GBA, SNCA, LRRK2, and MAPT. In addition, PD-causal monogenic variants account for 5 to 10% of cases [35, 37].
The severity and risk associated with GBA depend on the variant [38]. Overall, the GBA motor phenotype resembles idiopathic PD, possibly with faster progression, more bradykinesia, and levodopa-induced dyskinesias [39, 40]. Cognitive changes appear earlier and tend to be more prominent, particularly in memory and visuospatial domains [41–44]. The dementia risk with a severe GBA variant is 2.9 times higher than for mild variants and 5.6 times higher than for idiopathic PD [38]. Severe GBA variants may also have more neuropsychiatric and autonomic disturbances [45]. Bi-allelic carriers have faster disease progression and higher mortality than idiopathic PD [42, 47].
Autosomal dominant (AD) PD includes SNCA, LRRK2, and VPS35. LRRK2 variants are the most frequent cause of monogenic PD, with p.G2019S, p.R1441C, and p.R1441G being the most common. The phenotype resembles idiopathic PD, with atypical signs rarely reported [48]. A more uniform disease course regardless of the age of onset [51] and a higher likelihood to develop dystonia and dyskinesias earlier have been reported [49]. Like iPD, LRRK2-PD most likely manifests PIGD phenotype with disease progression [50]. Remarkably, LRRK2 p.G2019S carriers with a PIGD phenotype have a lower risk of dementia than observed in non-carriers with this phenotype [51]. Overall, LRRK2-PD have a lower risk of dementia [48, 52–54], manifests less olfactory impairment [49, 55], and RBD [56, 57] than non-carriers with PD.
SNCA variants include duplications or triplications and missense mutations, with p.A53T as the most frequent. Some SNCA variants (p.A53T and p.E46K) are more likely to develop dementia [58]. Depression, psychosis, and autonomic compromise are also more common for certain SNCA variants compared to idiopathic PD [5, 59–62]. For VPS35, the most common point mutation p.D620N presents with classical PD features with minimal atypical signs, although postural instability and daytime sleepiness may be more common [48, 64].
Autosomal recessive (AR) PD, such as PRKN, PINK1, and DJ1, present with a phenotype similar to idiopathic PD but with younger age of onset. Patients with PRKN variants can respond dramatically to low doses of dopaminergic agents [65]. In addition, it can present with exercise-induced lower extremity dystonia [66] and gait compromise associated with diphasic dyskinesias [67]. Variants of these three genes are uncommonly associated with atypical parkinsonian features [68].
On the other hand, ATP13A2, PLA2G6, FBXO7, DNAJC6, SYNJ1, and VPS13C represent AR forms that often manifest with juvenile parkinsonism, faster progression, and atypical features including supranuclear gaze palsy, oculomotor or eyelid apraxia, intellectual disability, facial-faucial-finger mini-myoclonus, ataxia, dysarthria, dysphagia, pyramidal signs, seizures, psychosis and dysautonomia [69–77].
Because of scarce evidence, how these causal or risk-modifying variants affect outcomes is debatable. Currently, decision-making for advanced therapies is based on clinical features, which are unreliable for inferring the underlying genetics. Incomplete penetrance (i.e., LRRK2, GBA), phenotype variability, and environmental factors affect clinical features [34]. At the same time, there is evidence that genetics affect outcomes in ways that would affect decision-making. This is reviewed in the next section.
CURRENT EVIDENCE FOR DEEP BRAIN STIMULATION IN MONOGENIC PARKINSONISM AND GBA VARIANTS CARRIERS
Pal et al. analyzed the Consortium On Risk for Early-onset PD (CORE-PD) cohort, emphasizing PRKN, LRRK2, and GBA. Ninety-nine individuals who received DBS, and 684 without DBS, were identified. Carriers of pathogenic (or “risk” variants for GBA) were more common in the DBS vs. non-DBS groups (26.5% vs. 16.8%, respectively) [78]. Performing genetic screening in a cohort of 94 DBS-treated PD patients, Angeli et al. found that 26% had PRKN, LRRK2 p.G2019S, or GBA variants. No pathogenic variants were found in SNCA [9]. Likewise, De Oliveira et al. reported that in addition to GBA variants, PRKN and LRRK2 were the most common monogenic forms in DBS cohorts [79]. Interestingly, the response to DBS seems to be related to the variants. Tables 2–5 summarize the evidence for outcomes obtained with device-aided therapies in monogenic parkinsonism and GBA carriers.
Available evidence of outcomes for different device-aided therapies in GBA variants carriers
LEDD, levodopa equivalent daily dose.
Available evidence of outcomes for different device-aided therapies in autosomal dominant monogenic parkinsonism
NR, not reported; LEDD, levodopa equivalent daily dose.
Available evidence of outcomes for different device-aided therapies in monogenic autosomal recessive parkinsonism
NR, not reported; LEDD, levodopa equivalent daily dose.
Available evidence of outcomes for different device-aided therapies in monogenic autosomal recessive parkinsonism presenting with atypical feature
NR, not reported; LEDD, levodopa equivalent daily dose.
Various authors have proposed different categories for motor outcomes. In their systematic review, de Oliveira et al. defined a mean UPDRS-III change of 50% or more as a marked response, a mean change of 30% to 50% as a satisfactory response, and less than 30% change as an unsatisfactory response [79]; on the other hand, Kuusimäki et al., defined an improvement of 30% or more in the UPDRS-III score as a favorable outcome; 20–30% a moderate outcome; and < 20% a poor/mild result [80].
DBS in carriers of genetic variants that modify the risk for developing PD or influence PD-related outcomes (GBA)
GBA carriers have DBS earlier in the disease course compared to LRRK2, PRKN, or non-mutation carriers [9, 78]. Most GBA carriers have marked or satisfactory short-term (<2 years) outcomes to STN-DBS [79]. Data for longer-term follow up are scarce, but outcomes tend to worsen over time. The authors hypothesized that because STN-DBS carries additional cognitive risk over GPi-DBS, the latter target may be preferable for GBA carriers, who are already at increased risk for cognitive impairment [79]. A separate study showed GBA carriers developed cognitive impairment and stimulation-resistant symptoms within 2 to 7 years after surgical treatment [81] (see Table 2). Thus, the overall benefit of DBS may be compromised due to the rapid progression of cognitive and neuropsychiatric symptoms [80].
A recent study screening for LRRK2, GBA, and PRKN mutations evaluating cognition at baseline and one-year post-DBS showed that high-risk or severe GBA variant was associated with pronounced postoperative cognitive decline [82]. The motor benefit was similar among groups.
Modeling different datasets, Pal et al. examined global cognition using the Mattis Dementia Rating Scale to compare the rate of change between GBA variant carriers and non-carriers with and without STN-DBS in PD. GBA carriers with DBS declined on average 2 points/year more than non-carriers with no DBS, 1.7 points/year more than GBA carriers with no DBS, and 1.5 points/year more than non-carriers with DBS [83]. Authors proposed that although non-randomized, this study suggests that GBA variants and STN-DBS’s combined effect negatively impact cognition, advising that GBA variant carriers should be counseled regarding potential risks associated with STN-DBS and alternative options may be considered [83].
Finally, the GPi target may be preferable for GBA carriers with dystonia and dyskinesia [79].
Both GPi-DBS and STN-DBS have similar outcomes on motor function measured by the UPDRS-III in the “on” and “off” medication state [84–86], and both targets have a beneficial effect on levodopa-induced dyskinesias [87]. STN-DBS achieves this goal mainly by a greater reduction in medication dosages [87, 88]; but also, stimulation of the area above the STN can directly suppress levodopa-induced “on”-dyskinesia [86]. In contrast, GPi-DBS may provide greater anti-dyskinetic effects possibly by a direct mechanism [84, 87]. Hence, clinical guidelines recommend GPi as the target, especially when reduction of medication is not anticipated, and there is a goal to reduce the severity of “on” medication dyskinesias [84, 89].
On the other hand, although it seems relatively safe concerning cognitive function, chronic stimulation of STN has been associated with a subtle decline in cognitive domains, exceptionally verbal fluency, and executive function [90, 91]. Despite little data is supporting that STN-DBS has a worse cognitive outcome than GPi-DBS [92], more published information is required for validation [93]; if there is significant concern about cognitive decline, particularly regarding verbal fluency, processing speed, and working memory in a patient undergoing DBS, GPi has been recommended [84, 89].
DBS in autosomal dominant PD (SNCA, LRRK2, VPS35)
A systematic review showed that LRRK2 p.R1441G had poorer outcomes than other LRRK2 variants [79]. Overall, the response of LRRK2 p.G2019S carriers to STN-DBS was comparable to idiopathic PD [81]. There are reports of another variant, p.T2031S developing neuropsychiatric problems 5–7 years after DBS [80].
Thus far in the literature, five individuals carrying a VPS35 p.D620N variant have undergone DBS (STN = 3, unreported target = 2) and were followed for 1 to 8 years. The motor outcome was favorable in 3, moderate in 1, and poor in 1 who developed gait impairment, dysarthria, behavioral changes, and cognitive decline a few years later [80].
In a meta-analysis of SNCA duplications, three individuals had bilateral STN-DBS with good results. Two did not have cognitive decline at four-year follow-up. However, the third individual developed dementia [94]. Another individual with mosaicism of SNCA duplication, with motor complications, mild cognitive impairment, hallucinations, and an impulse control disorder, had bilateral GPi-DBS eight years after symptom onset. Good motor benefit was reported 1 month after surgery [95] (Table 3).
In summary, outcomes appear favorable for the most common LRRK2 pathogenic variant, p.G2019S, but may be poor for p.R1441G due to rapid cognitive decline and worsening of neuropsychiatric symptoms. The evidence remains very limited for SNCA and VPS35, with heterogeneous outcomes.
DBS in autosomal recessive PD (PRKN, PINK-1, DJ1)
PRKN carriers tend to have earlier disease onset yet longer disease duration at DBS surgery [9, 78]. After DBS, most of them have sustained motor improvement and in activities of daily living comparable to idiopathic PD [81]. Data for GPi-DBS are scarce. One PINK1 homozygous patient had satisfactory motor improvement after STN-DBS, but long-term results are not available, and nonmotor outcomes were not described [96, 97]. We found no published data on DJ1 variants undergoing DBS (see Table 4).
DBS in autosomal recessive parkinsonism with atypical features
Bilateral GPi-DBS and ventralis intermediate nucleus (Vim)-DBS has been successfully utilized for dystonic storm treatment in a 15-year-old girl with atypical neuroaxonal dystrophy (NAD) phenotype, a subgroup of PLA2G6-associated neurodegeneration (PLAN). She had a complex clinical picture characterized by progressive generalized dystonia, spasticity, myoclonus, intentional tremor, oculogyric crises, seizures, and poor cognition. She achieved good control of dystonic storm symptoms, oculogyric crises, and tremors at a 9-month follow-up [98]. The use of DBS for the PLA2G6-associated dystonia-parkinsonism phenotype has not been reported.
A female carrier of a FBXO7 homozygous variant with juvenile parkinsonism associated with postural instability, dysarthria, hypophonia, marked motor fluctuations, and levodopa-induced dyskinesias was reported to have satisfactory motor control with multiple device-aided therapies. At age 21, five years after symptoms onset, CSAI reduced daily “off” time by 50%. However, within 6 months, the patient developed severe “on”-period generalized chorea-dystonic dyskinesias. Bilateral GPi-DBS was implanted and achieved good control of those symptoms at a 6-month follow-up. However, severe dysarthria progressed to permanent anarthria [99].
In a cohort of early-onset sporadic or familial PD, a 46-year-old homozygous DNAJC6 p.T741 = female carrier with levodopa-responsive parkinsonism since age 31, who developed severe motor complications, underwent bilateral STN-DBS with marked improvement. However, the follow-up time and details were not reported [100].
A Caucasian woman with parkinsonism since age 39 had severe dyskinesias under dopaminergic treatment, dysarthria, tremor, mild dementia, hallucinations, dystonia, gait, and gastrointestinal tract problems. She had compound heterozygous canonical splice-site variants in VPS13C (p.Lys639AspfsTer14, p.Leu678GlufsTer26, p.Ala1072GlufsTer14, p.Ala1072_Gln1110del, p.Ser1076ArgfsTer4). She had initial benefit from STN-DBS but developed severe dysarthria and mild aphasia after 2.5 years. Rapid progression of symptoms was reported at a 4-year follow-up [101].
A Persian male bearing a p.R449Q heterozygous mutation in ATP13A2, who was also known to carry two Parkin mutations—a deletion of exons 3 and 4 and duplications of exons 7 to 12, was reported to have parkinsonian symptoms, including rest tremor and a good response to levodopa, since the age of 36. At 50, a favorable response to STN-DBS stimulation was reported, with mild postural instability and depression but no atypical neurological signs [102].
EVIDENCE ON THE USE OF LEVODOPA CARBIDOPA INTESTINAL GEL
Autosomal dominant PD and GBA mutations
In a cohort of 12 PD patients on LCIG in the UK, the authors reported one patient with LRRK2. This carrier had a 19-year history of PD and died 24 months after LCIG initiation because of colon cancer [103]. In a study in Tel Aviv, where 44 PD patients underwent LCIG, five were LRRK2 carriers, four were GBA heterozygotes, two were GBA homozygotes, and another was a carrier of both GBA and LRRK2. No significant differences were found between the carrier versus non-carrier group [104]. The same study group published an abstract of the data from 69 Ashkenazi Jewish patients with known GBA (11 cases) or LRRK2 p.G2019S mutations (16 cases) and 42 idiopathic PD. Motor UPDRS scores were significantly higher, and levodopa equivalent daily doses (LEDD) were lower among GBA-PD than in the two other groups. Although not statistically significant, GBA-PD had a higher rate of hallucinations and lower cognitive scores. The latter could explain the lower LEDD in this group [105] (see Tables 23).
Autosomal recessive PD with homogeneous presentations
A juvenile PD patient carrying a PRKN p.T240M variant in a heterozygous state had a marked improvement in motor and non-motor scores on LCIG in a long follow-up period [106]. A 63-year-old PRKN carrier with a history of 35 years of parkinsonism died 3 months after introducing LCIG from unspecified causes [103].
A woman with homozygous PINK1 variants with parkinsonism since age 29 underwent LCIG at age 55 for motor fluctuations and dyskinesias. For three years, the motor response was satisfactory, but she required B6 vitamin replacement for sensory axonal polyneuropathy. Four years after LCIG, she developed marked dyskinesias, dopamine dysregulation syndrome, ICD, and punding [107].
EVIDENCE FOR CONTINUOUS APOMORPHINE SUBCUTANEOUS INFUSION
Autosomal dominant PD
In a case series of British LRRK2 patients, a 42-year-old man was reported to have a CSAI pump 14 years after disease onset for severe drug-induced dyskinesias. Before CSAI, he had depression, obsessive and hypomanic behavior, excessive alcohol drinking, and dopamine dysregulation syndrome. Outcomes were not specified [108].
Autosomal recessive PD with homogeneous presentations
Two PRKN patients have been reported with CSAI. The first case was a 32-year-old man with 25 years of progressive parkinsonism-dystonia syndrome, with deterioration of laryngeal dystonia on levodopa and severe neuropsychiatric symptoms. No outcomes were reported. The second case was a 57-year-old with a previous thalamotomy started on CSAI at age 47 with benefit. However, he developed psychosis with paranoid delusions that resolved after stopping apomorphine [109].
Autosomal recessive parkinsonism presenting with atypical features
A Turkish woman with homozygous PLA2G6 p.R747W and heterozygous LRRK2 p.W1295R variants developed parkinsonism at age 27. By age 29, she had severe parkinsonism with bilateral tremors, hypophonia, dysarthria, postural instability, and cognitive impairment. She had a good response to combined antiparkinsonian medications but developed irritability, restlessness, and ICD. By age 30, she was unable to stand or walk independently. After CSAI (5 mg/hour), she could walk for at least a 1-year follow-up. She developed intermittent visual hallucinations [110].
As mentioned above, a female FBXO7 homozygous variant carrier with juvenile parkinsonism was reported to have a transient motor benefit with CSAI. After 6 months, the patient developed severe “on” dyskinesias, requiring bilateral GPi-DBS [99].
DISCUSSION
The cumulative evidence for device-aided therapies in monogenic-PD and GBA carriers is still scarce.
Along with a regional difference in the prevalence of specific variants, the availability of advanced therapies is critical. Device-aided therapies offered in different countries may vary through healthcare systems, local experience, and center preferences. For instance, we have observed that publications on infusion therapies (i.e., LCIG and CSAI) in monogenic parkinsonism come predominantly from the UK and Middle Eastern countries (Israel, Saudi Arabia, and Turkey), and LCIG in GBA-PD from Israel. On the other hand, DBS-related publications are more widely distributed (i.e., North America, Europe, Middle East, Asia, South America, Australia), possibly because of increasing access to this therapy. Unfortunately, decision-making on device selection is not explicit in most reports from countries where more than one device-aided therapy is available (e.g., Italy, UK, Israel, Turkey, USA). Future reports should explain the selection of a specific device-aided therapy, especially when other alternatives are available.
Systematic reviews and a meta-analysis constitute the best evidence for DBS in monogenic PD. However, these are limited by small sample size, short follow-up, and incomplete data.
Moreover, several investigators have used different categories and cut-off values when defining DBS responses in gene-related PD populations; because of this heterogeneity, the same percentage of change in UPDRS-III would be qualified differently by distinct authors. An explicit limitation of this approach is the lack of consensus, adding difficulties when interpreting the literature. In addition to arbitrariness in establishing cut-off values, the effectiveness of these therapies has been firmly focused on the change in UPDRS-III scores, in our opinion lacking adequate emphasis on non-motor symptoms or changes in quality of life, which can be decisive in decision-making and in establishing the benefits of these therapies. Further, with some exceptions, reports on LCIG or CSAI lack objective and detailed results making a similar analysis difficult.
Mutation carriers seem to be overrepresented in DBS-cohorts compared to non-carrier PD populations. LRRK2 p.G2019S, homozygous or compound heterozygous PRKN, and GBA were the most frequent variants. This may be attributed to an overlap of the phenotype with criteria for device eligibility. However, this is not necessarily equivalent to suitableness in the selection. For instance, these all had motor outcomes comparable to patients with idiopathic PD, at least in the short term, with STN-DBS as the most frequent target. However, the motor benefit of STN-DBS in GBA-PD may be countered by a faster cognitive decline and axial symptoms following DBS than non-carriers. On the other hand, even if carriers may have poorer outcomes than non-carriers, this is not equal to absent benefit (e.g., motor benefit vs. cognitive decline). Future randomized studies should consider the quality of life as a primary outcome to better understand the risk-benefit ratio in GBA-PD.
This should be kept in mind when discussing prognosis, timing, and expectations for DBS.
Publications on DBS in autosomal recessive variants with atypical features are mainly limited to individual cases. Some patients have reported benefits, but outcomes are incompletely reported, and long-term data is scarce. Dysarthric speech, swallowing disturbances, freezing of gait, and balance problems are frequent features of atypical autosomal recessive parkinsonism (e.g., ATP13A2, PLA2G6, FBXO7, DNAJC6, SYNJ1, and VPS13C). On the other hand, these symptoms can occur using DBS parameters optimal for improving tremor, rigidity, and bradykinesia. Therefore, data is insufficient to differentiate device therapies outcomes (i.e., adverse effects) from disease progression or therapy non-responsiveness. At this point, in select cases, DBS seems to be a reasonable palliative therapy or a rescue treatment in emergencies such as dystonic storms.
Small genetic screening studies are the primary source of evidence for LCIG. There is no significant difference in motor outcomes between LRRK2 p.G2019S or GBA-carriers and non-carriers. GBA carriers in the LCIG studies had lower cognitive scores and higher scores for hallucinations.
CSAI has the most limited evidence of the three therapies in monogenic PD and GBA carriers. As expected, available cases tend to include individuals with very advanced diseases, given the typical patient selection criteria. CSAI may be a helpful alternative in recessive parkinsonism with atypical features, with some efficacy, as shown in the FBOX7 and PLA2G6 case reports.
The available information regarding individual monogenic variants and device-aided therapies is far from comprehensive. The data are limited to small numbers of patients, short follow-ups, and observational reports. Multicenter prospective cohort studies are needed to guide our knowledge and improve decision-making for device-aided therapies and PD-related variants.
In addition, we recommend that several key elements be included when reporting outcomes from device-aided therapy amongst genetic PD populations (Fig. 1).
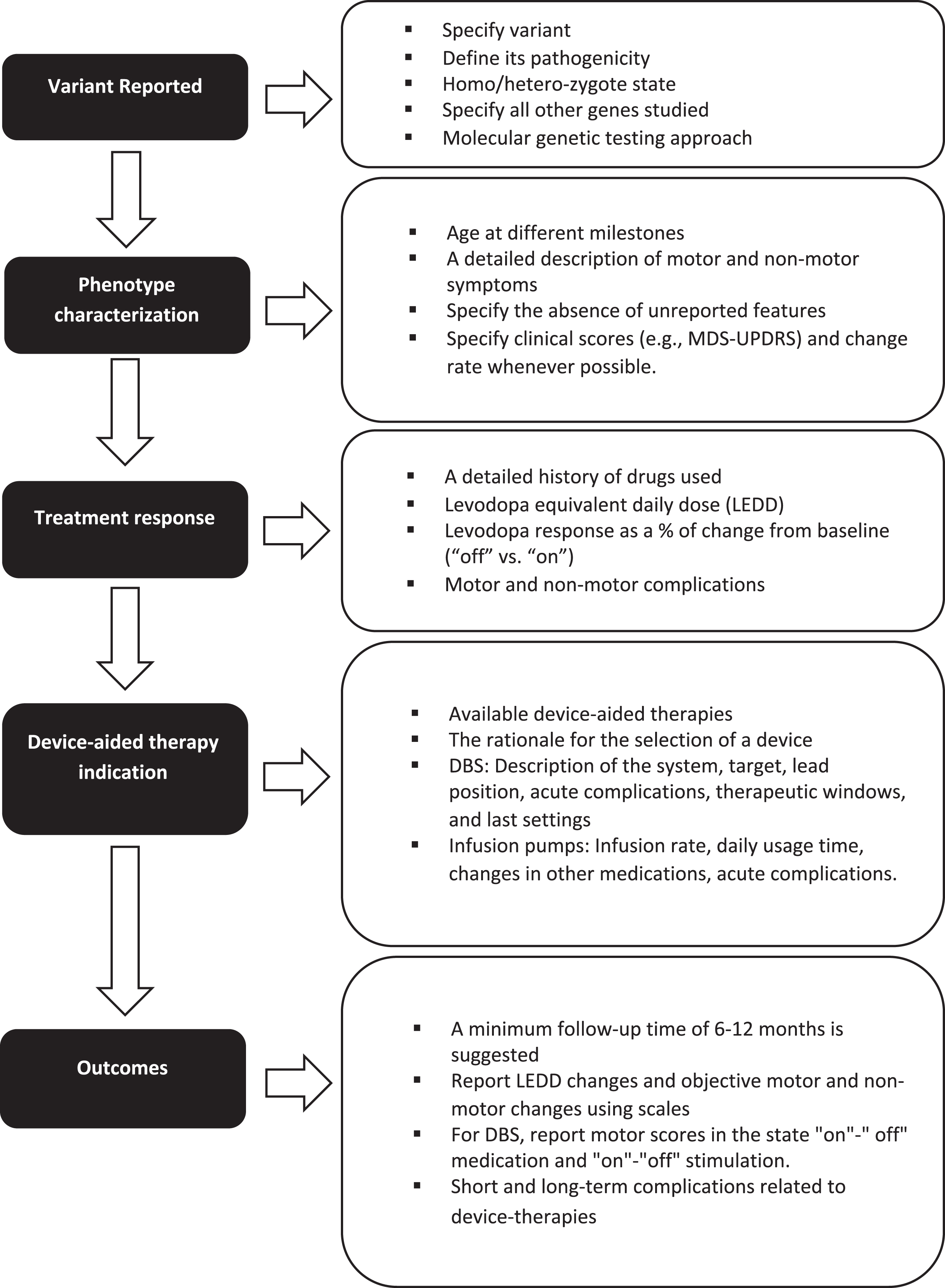
Key elements to consider when reporting the response to device-aided therapies in patients with monogenic parkinsonism or GBA variants carriers.
First, when discussing the genetic variant, the type of variant, its pathogenicity, and its homozygote or heterozygote state should be included. When using panels, all the genes studied should be mentioned, especially in patients belonging to ethnicities at risk for more than one type of variant (for example, LRRK2 p.G2019S and GBA variants in Ashkenazi Jews).
Second, when discussing phenotype, characterization must be rigorous, including the age at symptom onset, age at diagnosis, disease duration, initial clinical manifestation, presence of falls, freezing of gait, cognitive profile, neuropsychiatric manifestations, and other non-motor symptoms. The absence of unreported features should be specified. Individuals may be classified according to the MDS-UPDRS-III score (i.e., tremor dominant, intermediate, or postural instability/gait difficulty). Levodopa response should be described as % of change from baseline. Whenever possible, the rate of progression of motor and non-motor symptoms (i.e., cognitive decline) in the pre- and post-device-aided therapy stage should be included. A detailed history of the drugs used, related side effects and levodopa equivalent daily dose should also be included.
Third, the indication and rationale for each specific advanced device-aided therapy should be documented. In addition, for DBS, it is essential to define whether the surgery is uni- or bilateral, which commercial device was implanted, the target, lead position information, therapeutic window, and final stimulation parameters. The infusion rate, daily usage time, and changes in other medications should be indicated for infusion pumps.
Finally, long-term motor and non-motor outcomes should be measured objectively using, for example, the MDS-UPDRS scale administered at multiple time points. In the case of DBS, it is essential to report motor scores in the state “on"-” off” medication and “on"-“off” stimulation. Follow-up time should be sufficient for the device settings to reach a steady-state and assess disease progression, treatment efficacy, and long-term adverse effects. While there is no specific time, a reasonable minimum follow-up time would be greater than 6–12 months.
CONCLUSION
Based on current studies, it is unfeasible to establish evidence-based decision-making guidelines for device-aided therapies in monogenic parkinsonism. So far, an added prognostic value of genetic testing beyond a careful clinical assessment when patients are evaluated for device-aided therapies is yet to be demonstrated for monogenic parkinsonism. Large prospective cohorts combining genetic profiling with deep phenotyping, and randomized studies, can provide relevant data to address this question.
Although no randomized trials are available, based on accumulated evidence on the natural history and probable deleterious cognitive outcomes after STN-DBS in carriers of pathogenic variants in GBA, several authors have proposed that candidates for this device therapy should be screened for GBA mutations as part of the pre-surgical assessment. Carriers should be counseled regarding potential risks associated with STN-DBS, considering alternative options [83]. Comparative studies of different device-aided therapies in this population are pending.
We call for the development of guidelines that allow us to improve the quality and number of reports and randomized clinical studies that optimize our decision-making on device-aided therapies in monogenic parkinsonism and GBA carriers.
CONFLICT OF INTEREST
Grants/Research Support: Dr. Mata has received research support from American Parkinson’s Disease Association, Parkinson’s Foundation, Michael J. Fox Foundation, and NIH/NINDS
Grants/Research Support: Dr. Fernandez has received research support from Acorda Therapeutics, Alkahest, Amneal, Biogen, Michael J. Fox Foundation, Movement Disorders Society, NIH/NINDS, Parkinson Study Group, and Sunovion but has no owner interest in any pharmaceutical company.
Honoraria: Dr. Fernandez has received honoraria from Cleveland Clinic and Boston University as a speaker in CME events. Dr. Fernandez has received honoraria from Bial Neurology, Biopas, Cerevel, CNS Ratings, Denali Therapeutics, Kyowa Hakko Kirin, Pfizer, Partners Healthcare System, Parkinson Study Group, Revance, Sun Pharmaceutical Industries, Sunovion Research, and Development Trust as a consultant, and from Elsevier as the Co-Editor-In-Chief of Parkinsonism and Related Disorders Journal.
Royalty: Dr. Fernandez has received royalty payments from Demos Publishing and Springer for serving as a book author/editor.
Contractual Services: The Cleveland Clinic has a contract with Teva for Dr. Fernandez’s role as a Co-Principal Investigator in Deutetrabenazine for Tardive Dyskinesia global studies.