
Abstract
Parkinson’s disease (PD) is increasingly recognised as a systemic disorder in which inflammation might play a causative role rather than being a consequence or an epiphenomenon of the neurodegenerative process. Although growing genetic evidence links the central and peripheral immune system with both monogenic and sporadic PD, our understanding on how the immune system contributes to PD pathogenesis remains a daunting challenge. In this review, we discuss recent literature aimed at exploring the role of known genes and susceptibility loci to PD pathogenesis through immune system related mechanisms. Furthermore, we outline shared genetic etiologies and interrelations between PD and autoimmune diseases and underlining challenges and limitations faced in the translation of relevant allelic and regulatory risk loci to immune-pathological mechanisms. Lastly, with the field of immunogenetics expanding rapidly, we place these insights into a future context highlighting the prospect of immune modulation as a promising disease-modifying strategy.
INTRODUCTION
Heterogeneous and multifactorial in nature, Parkinson’s disease (PD) follows a complex model of inheritance spanning the etiological spectrum ranging from monogenic disease (in a small proportion of affected individuals) to polygenic inheritance (in the vast majority of the cases) where environmental and genetic risk factors interact to induce PD pathology.
Emerging and compelling evidence supports chronic neuroinflammation, derived from impaired innate and/or adaptive immunity mechanisms, as among the main contributors to PD development, in which a pro-inflammatory state may trigger or promote neuronal loss [1]. A growing body of research recognises PD as a systemic disease characterised by the presence of central and peripheral inflammatory processes.
In recent years, extensive research in the PD genetics field has focused on unravelling both coding and non-coding genetic variation that could result in immune defects contributing to PD risk, onset, and progression. Indeed, the advent of high-throughput, high-resolution next-generation sequencing and genome-wide genotyping technologies has been instrumental in the identification of genes and regulatory genomic regions that play a prominent role in immune processes leading to the pathophysiology of the disease.
This review aims to provide an updated overview of the role of monogenic PD genes in the regulation of the immune system as well as our current knowledge on the contribution of recently identified immunogenetic risk factors leading to neurodegeneration in PD. We outline the challenges faced in the translation of relevant allelic and regulatory risk loci to immune-pathological mechanisms and discuss potential shared genetic etiologies between PD and autoimmune diseases. Furthermore, we illustrate the increasing need to generate harmonised large-scale omics, clinical and longitudinal data to enhance our understanding of the immune system involvement in PD etiology that could aid future development of immunomodulatory therapeutic interventions aimed to prevent or delay disease onset.
THE ROLE OF MONOGENIC PARKINSON’S DISEASE KNOWN GENES IN THE REGULATION OF THE IMMUNE SYSTEM
Monogenic PD —PD caused by a defined mutation in a single gene—constitutes about 5–10% of the total PD cases [2]. Mutations in several genes have been identified as causative for monogenic PD with different patterns of inheritance including autosomal dominant, autosomal recessive, and X-linked [3]. Scrutinising the molecular processes through which mutations in such genes culminate in the development of PD, modulation of immune-related pathways emerged as a potential pathogenic mechanism [4, 5]. This has stirred research to dissect the potential roles of monogenic PD-known genes in regulating the immune response [6].
LRRK2
The leucine-rich repeat kinase 2 (LRRK2) gene has been linked to monogenic as well as sporadic PD. LRRK2 mutations are the most frequently reported genetic factors in monogenic PD, representing 5–6% of familial PD cases and 1–2% of sporadic PD with variability in prevalence between different populations [7, 8]. The incomplete penetrance of LRRK2 indicates the involvement of other genetic/environmental components, which might act modifying the pathogenic effects of such mutations [9, 10]. LRRK2 gene encodes a multi-function protein with kinase and GTPase activity that is found associated with PD pathogenesis. The structural homology of LRRK2 with receptor-interacting protein kinases, a family of kinases with known implications in regulating the immune system, highlights the potential role of LRRKs in regulating immune-related pathways that might contribute to PD pathogenesis [11]. This has been further supported by the fact that LRRK2 expression in peripheral immune cells (such as B-cell, T-cell, and monocytes) is markedly upregulated in PD with a coinciding upsurge in the activity of these immune cells [12]. Results from numerous studies advocate that LRRK2 modulates immune responses to favor sustained inflammation and subsequent neurodegeneration in PD patients. For instance, mutated LRRK2 was found to promote neuroinflammation through increasing microglia and macrophages chemotaxis to the brain tissues via modulating pathways controlling cell adhesion, polarisation, and directional motility. LRRK2 is also implicated in modulating processes like phagocytosis, and the production of proinflammatory cytokines [13, 14]. Furthermore, LRRK2 mediates such effects by stimulating receptors like TLR2 and TLR4, and through several downstream kinases, mediators, and cytokines [4, 15–17]. It is noteworthy that LRRK2 effects on the immune response are genotype dependent. This has been observed in mouse models where a gain-of-function mutation (p.G2019S) in the LRRK2 kinase protein resulted in enhanced inflammatory responses, while the kinase inactivating mutation (p.D1994S) produced an opposite effect.
Additionally, studies have shown that LRRK2 mutants contribute to neurodegeneration through exacerbating neuroinflammation via peripheral proinflammatory cytokines. According to recent reports, LRRK2 maintains neuroinflammation through stimulating peripheral immune cells type II interferon responses rather than effects on central microglia [18]. Within the same context, LRRK2 was proved to influence myelopoiesis and peripheral myeloid cell differentiation [19]. Effects of LRRK2 on peripheral immune responses and its implication in PD pathogenesis are currently under intense investigation to analyse the interplay between central and peripheral immune responses and decipher the link between PD and peripheral inflammatory conditions like Crohn’s disease (CD), ulcerative colitis (UC), and leprosy [20, 21].
SNCA
The SNCA gene encodes alpha-synuclein, the histopathological hallmark of PD. Several mutations and genetic rearrangements in SNCA have been linked to autosomal dominant PD [22, 23]. Moreover, the dosage of SNCA has been found closely associated with PD onset, clinical phenotype, and patient’s survival [24]. Given that earlier onset and more rapid disease progression is observed in patients with SNCA gene triplications as compared to those with duplications, a dosage effect of the SNCA gene is expected. These observations indicate a neurotoxic effect of increased alpha-synuclein, even when point mutations are not present. The expression of alpha-synuclein in different immune cells, and the reported colocalization of neuroinflammatory spots at the sites of alpha-synuclein aggregation (Lewy neurites/Lewy bodies) indicate a potential implication of immune-mediated mechanisms regulated by alpha-synuclein in the pathogenesis of PD [25]. Alpha-synuclein has been proved to impact both the innate and adaptive immune responses in a way that favours neurodegeneration and PD development. Acting as a ligand of TLR-2 and TLR4 receptors or via metabolic reprogramming, alpha-synuclein is responsible for launching a proinflammatory response of microglia leading to neuronal demise probably through the release of neurotoxic mediators or phagocytosis of the neuronal cells in the region [26, 27]. Degenerated neurons release intracellularly aggregated alpha-synuclein to the extracellular space leading to further immune stimulation and neurodegeneration, a cascade that eventually leads to PD development. The neuroinflammation can be further aggravated by the microglial and astrocytes major histocompatibility complex class-II (MHC-II) presentation of alpha-synuclein leading to the recruitment and activation of other peripheral phagocytes (such as monocytes, macrophages, and lymphocytes) [28, 29]. Similar inflammatory responses mediated by alpha-synuclein were reported in the enteric nervous system, a finding that implies a potential role of gastrointestinal (GI) infections or inflammatory conditions in PD pathogenesis [30].
Moreover, alpha-synuclein has evident roles in regulating the magnitude and quality of adaptive immune responses in many aspects [31]. For instance, alpha-synuclein was found essential for the development and activation of humoral immunity, since SNCA knock-out mouse models demonstrated impaired B cells development and IgG production [32, 33]. In addition, alpha-synuclein activity is also important for T cell development and differentiation [32]. Deficiency of alpha-synuclein was accompanied by elevated IL2 and Th1 response and decreased IL4 and Th2 differentiation, a finding that further emphasises the role of alpha-synuclein in T cells differentiation and priming [32, 34]. Furthermore, in support of the notion that neurodegeneration in PD might be induced by autoimmune responses, it has been observed that alpha-synuclein induces substantia nigra (SN) neurons to display major histocompatibility complex class-I (MHC-1) presenting immunogenic alpha-synuclein epitopes that are recognized by T cells [35, 36]. Such alpha-synuclein reactive-T cells have been linked to the development of PD and are now proposed as early diagnostic markers of the disease [37].
VPS35
Mutations in the vacuolar protein sorting 35 ortholog (VPS35) gene have been reported in about 1% of monogenic PD and 0.2% of sporadic PD [38]. The VPS35 gene that has been linked to late-onset, autosomal dominant familial PD, encodes a core component of the retromer complex responsible for protein transmembrane sorting through the endosomes -trans-Golgi network pathway [38, 39]. VPS35 is expressed in neuronal immune cells, namely microglia, and astrocytes. Recent studies have reported the role of VPS35 in regulating innate system responses through modulating microglia and astrocytes activation [40, 41]. VPS35 regulates microglial functions and polarisation through modulating the trafficking and recycling of immunomodulating receptors/mediators. Scientific literature exploring the effects of VPS35 on microglia have been contradicting, probably due to variation in the brain anatomical sites and the inducer of neuroinflammation [41, 42]. One report indicated that VPS35 favours the priming of microglial response into a pro-inflammatory rather than an anti-inflammatory response following an ischemic brain injury in the brain cortex [41]. On the contrary, in vitro and murine model studies proved that VPS35 deficiency is associated with an enhanced microglial activation and excessive neuroinflammation through induction of microglial inflammatory mediators in the hippocampus [42, 43]. Furthermore, recent studies suggest that VPS35 interacts with LRRK2. It has been demonstrated that LRRK2 phosphorylates some RAB proteins, which are involved in vesicle trafficking, [44] and the D620N VPS35 mutation is implicated in hyper-activation of LRRK2 kinase pathway [45].
PRKN, PINK1, DJ1
The Parkin RBR E3 ubiquitin-protein ligase (PRKN), PTEN-induced kinase 1 (PINK1), and DJ1 (also known as PARK7) genes are widely known to contribute to autosomal recessive PD and encode proteins that are essential regulators of mitochondrial homeostasis and quality control [46]. PINK1 and PARKIN control mitochondrial turnover via a selective autophagy process known as mitophagy [47]. They are also involved in regulating mitochondrial fusion and fission, local repair of impaired mitochondria, and the genesis of new mitochondria [48]. DJ1 works interactively with PARKIN and PINK1 in the mitochondrial quality control through regulating oxidative stress caused by ROS [49]. The three genes, which are known for their neuroprotective roles, are linked to recessive PD with their mutations reported in 13% of early-onset PD. The similarity in disease phenotype and clinicopathological features suggests similar pathogenic pathways through which the three genes culminate in PD development. Among the reported pathogenic mechanisms through which PRKN, PINK1, and DJ-1 genes contribute to PD is neuroinflammation. Neuroinflammation caused by loss-of-function mutations of these proteins can be mitochondrial-mediated, e.g., impaired mitophagy caused by PARKN/PINK1 deficiency results in the release of mitochondrial DNA and other reactive species that can stimulate innate immunity and trigger inflammation [50, 51]. Furthermore, PRKN/PINK1 loss of function mutations impair the proteins inhibitory effect on mitochondrial antigen presentation through the alternative mitochondrial-derived vesicles (MDV) pathway leading to excessive mitochondrial antigen presentation and stimulation of immune responses according to studies conducted in in vitro and in vivo models [52]. The unleashed mitochondrial antigen presentation through the MDV pathway caused by the loss of PRKN/PINK1 regulatory effect further supports the autoimmune basis of PD. It also supports the role of the brain/gut axis in PD pathogenesis, considering that PINK1-/- mouse models have demonstrated a propagated inflammatory response after bacterial infection with concomitant development of peripheral and neural cytotoxic mitochondrial-specific CD8 + T cells. The consequent decline in dopaminergic neuron density and development of motor impairment in the mice indicate that the PINK1-/- associated autoimmune response likely contributes to dopaminergic neurodegeneration and PD development [52]. Moreover, PRKN/PINK1 deficiency can lead to an exacerbated inflammatory response through inhibition on the NLRP3 inflammasome signalling [53, 54]. Similar to PRKN/PINK1 mutations, DJ-1 dysfunction was observed in CD4 + T cells to enhance neuroinflammation due to the loss of DJ-1 antioxidant effect and regulatory ROS mediated inflammation [55].
Furthermore, these proteins can also induce neuroinflammation through other mitochondrial-independent mechanisms, e.g., PINK1 deficiency was reported to promote innate immune responses through nitric oxide production by glia/astrocytes cells leading to inflammation-induced neuronal death [56]. Additionally, DJ-1 exerts anti-inflammatory effects independently from the ROS regulatory pathways, e.g., through inducing the synthesis of anti-inflammatory mediators like prostaglandin D2, stimulating the migration of CD3 + T cell, and CD4 + T cells differentiation [57, 58].
GBA
Mutations of the glucocerebrosidase gene (GBA), encoding for the lysosomal enzyme β-glucocerebrosidase, are the most prominent genetic risk factor recognized for PD. According to the records, GBA mutations, which are detected in 8–12% of total sporadic PD in the world, increase the risk of developing PD by 5–10 folds in a carrier compared to the general population [59]. GBA is expressed in immune cells including monocytes/macrophages and lymphocytes, and it has been associated with an aberrant inflammatory response mediated by monocytes/macrophages and B cells [60, 61]. In addition, the glucocerebrosidase enzyme activity is generally reduced in monocytes from PD patients compared to control cases according to published reports. Such reduction was found correlated with the disease’s clinical characteristics, which highlights the potential of peripheral monocytes glucocerebrosidase activity as an early marker of PD diagnosis [62, 63].
Moreover, GBA mutations demonstrated association with marked astrocytes and microglial activation indicating that GBA mutations are implicated in launching and/or aggravating neuroinflammatory responses [64, 65]. Such effect is detected early enough before neurodegeneration, a time where therapeutic intervention can be maximally beneficial. Hence, investigating the mechanisms and mediators through which GBA mutations initiate and propagate neuroinflammation is now a hot area of research [66]. Despite the mentioned association of GBA mutations with central and peripheral inflammatory responses, initial investigations of central and peripheral inflammatory cytokines as early markers of PD have returned with negative outcomes [67].
COMMON GENETIC RISK FACTORS CONTRIBUTING TO THE IMMUNE SYSTEM RESPONSE IN PARKINSON’S DISEASE
Since the development of advanced genotyping, sequencing technologies and complex analysis tools, large-scale genetic studies have expanded rapidly and have provided opportunities for a mechanistic elucidation of PD etiology. Since 2009, genome-wide association studies (GWAS) and meta-analyses have shown evidence for an implication of genetic contributors conferring moderate and low risk to PD susceptibility. The largest and latest GWAS meta-analyses conducted in Europeans, Asians, and Latino populations, have nominated a total of 92 loci predisposing to PD as well as several other suggestive genomic regions linked to age at disease onset and progression that warrant further study [68, 69]. PD risk loci such as LRRK2, MAPT, BST1, and HLA among others have been recognized as key players to the immune-mediated response involved in PD development (Table 1) [21], strengthening the hypothesis that common variation plays a crucial role in disease etiology. Furthermore, expression and methylation quantitative trait loci analyses have further nominated possible functional mechanisms by which genetic variants may be contributing to the immune system regulation [21].
The implication of PD risk loci in the immune system regulation
While GWAS meta-analyses have identified increasing numbers of novel genetic risk loci in myriad datasets, recent research in the PD genetics field has focused on understanding how genetic risk variants may disrupt biological processes and drive the underlying pathobiology of the disease. In this context, an exciting era for PD research has arisen with large-scale omics analyses supporting the role of the immune response in the pathophysiology of PD [21, 71]. Based on an unbiased approach applied to large genetic and genomics datasets available to date, pathway-specific polygenic risk scores and transcriptomics analyses have identified that a cumulative effect of common genetic risk variants contribute to the innate and adaptive immune responses, as critical pathways in PD etiology [70]. In concordance with this study, heritability and gene-set enrichment methods aimed at exploring particular functional marks for regulatory activity and gene-set lists have supported the implication of the innate and adaptive immune system in PD etiology and an enrichment for sporadic PD genetic heritability [72].
Interestingly, a more recent study has shown a significant enrichment of PD risk heritability in microglia and monocytes [73]. As an effort to understand the contribution of the immune system in PD pathogenesis, the authors showed that the microglial signature gene, P2RY12, located near a PD GWAS signal colocalizes to a microglia open chromatin region and enhancer region [73]. P2RY12 has been found to be downregulated in Alzheimer’s disease (AD) brain sections of microglia as a marker of inflammation, hence supporting a possible similar role in PD as a targetable microglial gene candidate and pathogenic player [74]. Additionally, a recent genetic analysis of the human microglial transcriptome across brain regions, aging and disease pathologies has nominated microglia-specific enhancers, finding associations with microglial expression of USP6NL for AD and P2RY12 for PD [75].
Furthermore, much attention has been paid to explore the role of immune cells in PD pathogenesis from a genetics perspective. Interestingly, recent studies showed that genes within PD GWAS loci are specifically expressed in T-cells [72]. Of note, GWAS have nominated a transcription factor named SATB1, which is associated with T-cell function and the establishment of immune tolerance [76, 77]. Specifically, the number of infiltrated CD8 positive T-cells is elevated in the PD substantia nigra pars compacta which correlates to neuronal cell loss. In addition, half of CD8 positive T-cells express an immune activation marker named IFNγ [78].
Moreover, PD-associated variants, such as rs76904798 which regulates the expression of LRRK2, had been shown to colocalize with peripheral monocyte expression quantitative trait loci (eQTL) [71]. Recent publications supported the notion that LRRK2 levels were elevated in PD patient monocytes which tended to secret more inflammatory factors and cytokines than healthy subjects [12]. Although LRRK2 plays a regulatory role in immune cells and PD, the mechanism of LRRK2 regulating PD patient monocyte or T-cell gene expression has not been yet unravelled.
Besides immune responses nominated through polygenic risk of biological processes, additional pathways have also been shown to induce immune activation in neurodegenerative disease through common genetic variation, such as the alpha-synuclein pathway and the MAPK signalling pathway [70]. Previous studies demonstrated that microglia activation was induced by aggregated alpha-synuclein, and this phenomenon raised dopaminergic neurotoxicity [79]. An additional supportive study has shown that alpha-synuclein induced the inflammatory factor interleukin-6 (IL-6) through MAPK MEK1/2, JNK and p38 MAPK in human astrocytes [80]. Therefore, genetic variation in the SNCA locus may also be responsible for inducing innate and adaptive immunity [81, 82]. MAPKs are a group of serine/threonine protein kinases participating in the regulation of inflammatory mediator production, stress response and maintenance of the immune system, which contribute to PD pathology [83, 84]. In addition, MAPKs contribute to T-cell differentiation and maturation, as well as immune cell activation [85, 86].
The majority of nominated risk variants are located in intronic and intergenic regions, regulating gene expression. For example, rs1990622, a genetic variant thought to affect cognition in PD, has the ability to enhance long-range chromatin looping by promoting the interaction between the chromatin organising protein CCCTC-binding factor (CTCF) downstream of TMEM106B and distal regulatory elements that in turn act increasing the expression of TMEM106B [87, 88]. TMEM106B has been found to modulate inflammation in the central nervous system (CNS) of post-mortem aged brains [89, 90]. Whole-genome co-expression network analysis nominated five gene clusters derived from frontal cortex data analyses conducted in elder individuals and categorised CNS cell types where these clusters were more differentially expressed, including microglia, astroglia and other neuron-associated groups. Interestingly, the rs1990622 genotype showed a significant elevation in expression with increased risk allele load on the microglia-associated genes cluster [89]. Further analyses confirmed the relationship between microglia gene set expression levels and the rs1990622 risk variant [89]. In this study, authors demonstrated that TMEM106B modulates the polarisation of immune cells towards pro-inflammatory stage of gene expression signature in the risk allele (Thr185) compared with the protective allele (Ser185) of rs1990622 in human monocyte-derived dendritic cells [89]. This suggests that PD-associated SNPs participating in immune response or immune-associated genes might interact with CTCF binding sites, regulating gene expression in harmful neuronal cells, and increasing the risk of neurodegeneration. However, further verification and discussion are needed in PD studies.
Heritable alterations contributing to regulatory gene expression, such as DNA methylation and histone modifications, which alter chromatin accessibility and gene regulation have been linked to PD etiology. Large-scale analysis also indicated that chromatin remodelling and organisation which is highly integrated in maintaining and controlling chromatin landscape and genome stability play a crucial role in PD pathogenesis [70]. A recent study revealed that BIN1, SORL1 and MEF2C show differentially methylated enhancers in the prefrontal cortex of Alzheimer’s patients [91]. In addition, evidence supports that a portion of genetic variants contributes to active and repressive chromatin states, such as heterochromatin and euchromatin [92]. Epigenetic regulation contributing to the immune system regulation affords an opportunity to develop potential therapeutic strategies in PD as neuroprotective targets in disease onset and progression (Fig. 1).
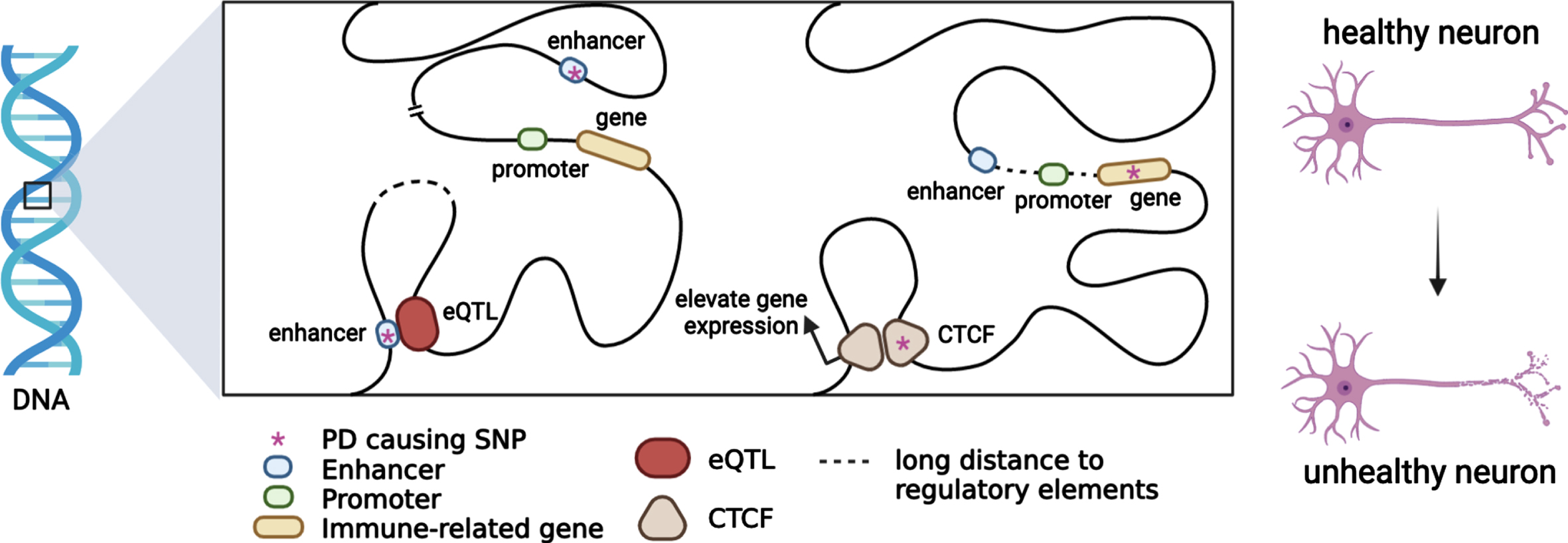
Putative functional mechanisms for which PD genetic variants might contribute to the immune system regulation. Most PD risk loci span non-coding regions. When a PD risk SNP with an eQTL mechanism colocalizes with immune-related genes in topologically associating domains, immune-gene regulation will be affected by chromatin interaction. PD causing SNPs may be located in enhancers or promoters, and immune-gene regulation will be affected by trans-acting or cis-acting regulatory mechanisms.
GENETIC COMORBIDITIES BETWEEN PARKINSON’S DISEASE AND AUTOIMMUNE DISEASES
Several genetic factors have been found to be common between PD and autoimmune disorders. For instance, genome-wide analysis identified 17 shared loci adjacent to GAK, HLA-DRB5, LRRK2, and MAPT genes for PD and 7 autoimmune diseases, including rheumatoid arthritis, UCs, and CD, suggesting that moderate and low risk loci contribute to pleiotropy among these diseases [21].
The Human leukocyte antigen (HLA), also known as major histocompatibility complex (MHC) is a family of genes encoding membrane proteins responsible for antigen presentation to T cells, hence playing a crucial role in the adaptive immune response [93] and it is the most reported genetic risk factor associated with autoimmune diseases and has been widely linked to several neurological conditions [94]. The HLA region was first identified to be associated with late onset PD in 2010 and is commonly referred to as the PARK18 locus [95]. More recently, fine-mapping analyses have nominated four HLA types as associated with PD: HLA-DQA1*03:01, HLA-DQB1*03:02, HLA-DRB1*04:01, and HLA-DRB1*04:04 [96]. Furthermore, identification of new SNPs has raised the of PD having an association with regulatory elements effecting the HLA class II gene expression and classical HLA class II allele [97]. Since then, different approaches have been undertaken to further dissect the role of HLA in PD and autoimmune conditions.
Genetic characterization of the HLA-DRB1 locus have revealed shared genetic risk factors associated with rheumatoid arthritis (RA) and PD. Such association remains controversial since the directionality of effect for both diseases is sometimes inconsistent, suggesting either a predisposing (HLA-DRB1*01:01 allele) [98] or protective effect (HLA-DRB1*04 allele) [98, 99]. Therefore, there is an ongoing debate about the direction of the association between PD and RA [100]. In an attempt to further clarify such an association, research aimed at exploring genetic correlations between these diseases has been conducted in European and non-European populations. In a Taiwanese case-study, PD incidence was observably higher in RA patients than in controls [101] and a significant genetic correlation between PD and RA has been reported [102]. More recently, a Mendelian randomization study showed a genome-wide negative correlation, suggesting that RA has a protective effect on PD [103], a conclusion supported by other epidemiological case studies [104].
Likewise, the etiology of CD, an inflammatory bowel disorder, is thought to be partly explained by the contribution of variants in the LRRK2 gene, which is considered among the main genetic contributors to PD risk [105]. Exome sequencing and genotyping of CD cases and controls has recently suggested that the N551K variant in LRRK2 provides protection from CD, while the N2081D variant confers risk, reinforcing the notion that both protective and harmful influences contribute to the complex genetic architecture of immune contributors [20]. Additionally, one of the first identified shared polymorphisms between CD and PD was M2397T [106] which presumably enhances interferon-γ responses in monocytes thus modifying the immune response [107]. Besides LRRK2, other genes have also been studied in CD/PD. The Solute Carrier Family 39 Member 8 (SLC39A8) gene encodes for a membrane manganese transporter protein, and the missense variant A391T has been previously linked to both CD [108] and PD [109].
Additional candidate shared genes between PD and CD have recently come to attention. There are hints that both alpha-synuclein and tau proteins, encoded by the SNCA and MAPT genes respectively, and considered pathological hallmarks in PD, may also be involved in CD: increased expression levels of alpha-synuclein protein were detected in inflamed tissues of CD patients [110] and tau protein expression was found upregulated in the enteric nervous system in CD patients [111]. However, these studies were carried out at a protein level and do not provide enough evidence to assess genetic comorbidity.
Epigenetic changes have also been investigated between PD and autoimmune diseases. Estimation of Pearson’s correlation coefficient between PD and RA revealed that both traits share 337 gene pairs with co-methylation changes [112]. Also, a significant overlap in epigenetic patterns of risk genes for PD, RA, and CD was found in butyrate-associated methylation sites [113]. Investigating PD from an epigenetic angle may clarify the role of immune response and immune-mediated mechanisms.
FUTURE DIRECTIONS
Increasing evidence from recent literature suggests that PD presents genetically as more of a systemic disorder in which inflammation might play a causative role rather than being a consequence or an epiphenomenon of the neurodegenerative process. As discussed, this notion is reinforced by the overlapping genetic pleiotropy that seems to exist between PD and several autoimmune diseases. Additionally, genes implicated in monogenic forms of PD and genome-wide loci linked to idiopathic PD have been seen to be widely implicated in immune system related processes including antigen presentation, inflammation regulation and the complement system.
Despite efforts at dissecting genetic drivers and modifiers of PD etiology that modulate the immune response, gene-environment interactions merit further investigation. Neuroinflammation is triggered by immunological insults likely at the convergence of genetic and environmental factors for which the exact mechanisms of such association remain to be elucidated. The challenge ahead lies in our ability to further dissect immune targets by defining the complex genetic architecture of the immune system contributors to disease risk, onset and progression, as well as its functional implications, and the interplay that exists with the environment.
The field of immunogenetics is rapidly expanding but there are still caveats to be addressed. Most of our current knowledge on the role of the immune system in PD etiology comes from human studies conducted in European populations. As we move forward, shedding light on immunogenetic contributors in Non-European populations and its interaction to specific environmental factors is essential to provide generalised insights into disease pathophysiology. Looking to the future of immunogenetics in PD, multimodal data integration with harmonised large-scale, clinica-longitudinal data and collaborative worldwide initiatives will facilitate translation of immunogenetic factors to specific functional mechanisms. Although much work needs to be done from the genetics and genomics perspective to inform biology, future immune-based therapeutic approaches aimed at modifying the expression of PD risk loci in the right patients and at the right time may succeed.
Footnotes
ACKNOWLEDGMENTS
This work was carried out with the support and guidance of the ‘GP2 Trainee Network’ which is part of the Global Parkinson’s Genetics Program and funded by the Aligning Science Across Parkinson’s (ASAP) initiative.
This research was supported, in part, by the Intramural Research Program of the National Institutes of Health (National Institute on Aging, National Institute of Neurological Disorders and Stroke: project numbers 1ZIA-NS003154, Z01-AG000949-02 and Z01-ES10198).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.