
Abstract
Electronic and structural features of some of representative chromene derivatives were investigated in this work towards recognizing their anticancer roles. Density functional theory (DFT) calculations were performed to obtain five structures of chromene derivatives with the same skeleton of original structure. In addition to obtaining optimized structural geometries, electronic molecular orbital features were evaluated for the models. Energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) indicated effects of additional R group pf chromene derivatives on electronic features. Based on such results, it was predicted that one of derivatives, L5, could better participate in interactions with other substances in comparison with other ligand structures. This achievement was obtained based on availability of HOMO and LUMO levels in lower energies easily catchable for electron transferring. On the other hand, L5 was assumed to interact in the weakest mode with other substances. Indeed, the main goal of this work was to examine anticancer activity of the investigated chromene derivatives, in which each of L1–L5 chromene derivatives were analyzed first to recognized electronic and structural features. Next, molecular docking (MD) simulations were performed to examine anticancer role of L1–L5 against methyltransferase cancerous enzyme target. The results indicated that formations of ligand-target complexes could be occurred within different types of interactions and surrounding amino acids of central ligand. In agreement with the achievements of analyses of single-standing L1–L5 compounds, L4-Target was seen as the strongest complex among possible complex formations. Moreover, values of binding energies and inhibition constant indicated that all five chromene derivatives could work as inhibitors of methyltransferase cancerous enzyme by the most advantage for L4 ligand. And as a final remark, details of such anticancer activity were recognized by graphical representations of ligand-target complexes showing types of interactions and involving amino acids in interactions.
Introduction
Cancer has been always a serious issue threatening the health system of human life all around the world without any certain solution up to now [1]. Several people have been dealing with cancer problem during recent centuries, in which the available protocols of medications were not suitable to save their lives [2]. Hence, numerous patients are victims of cancer every year [3]. Therefore, it is a must to explore new protocols for medication of cancer patients to save their lives or to increase their life quality [4]. Among invasive and non-invasive medical treatments, the non-invasive treatment is better to be employed not to destroy the living tissues of human body besides the cancerous tissues [5]. To this aim, performing medicinal chemistry research works could help to innovate novel pharmaceutical compounds for the purposes of medication of unknown diseases such as cancer [6–8]. It is also important to mention that developing techniques for recognition of cancer is also an important task regarding the early stage detection of cancer and other diseases [9, 10]. Computer-based tools have been developed in this case to investigate chemical compounds at the smallest molecular and atomic scales to find benefits of their use for different targets in living systems [11–13]. For innovating novel pharmaceutical compounds for medication of cancer, employing such computational techniques could help to provide insightful information about the investigated systems avoiding the existence of complexity of experimental environments [14–16]. For this purpose, several computational-based approach methodologies and techniques have been developed up to now [17–19].
In this work, such technical advantages have been used to compute electronic and structural features of some of chromene derivatives in addition to evaluating their anticancer activity. To this aim, density functional theory (DFT) calculations were performed to obtain optimized structures of representative chromene derivatives and their corresponding electronic and structural features. Next, molecular docking (MD) simulations were performed to examine anticancer activity of investigated structures against methyltransferase enzyme target. Earlier works indicated importance of performing such research works for exploring new inhibitors for the cancerous enzymes, in which the results have not been finalized yet [20–22]. Heterocyclic compounds have been always appropriate candidates for working as inhibitors against cancerous enzymes [23]. Chromene derivatives have also such advantages of heterocyclic compounds for possible inhibition of cancerous enzymes, in which further investigations are still required to approach the goals of drug design towards cancer issue [24, 25]. Evaluating electronic and structural features of compounds in terms of quantum descriptors could help to predict their characteristic roles for interacting with macromolecular targets [26–28]. Accordingly, such points have been examined in this work for some of chromene derivatives to analyze their electronic and structural features first and to evaluate their anticancer activity next, in which the required information for approaching this goal were summarized in Tables 1 and Fig. 1.
Molecular descriptions of the chromene derivatives.
Molecular descriptions of the chromene derivatives.

Molecular docking results of ligand-target interactions.
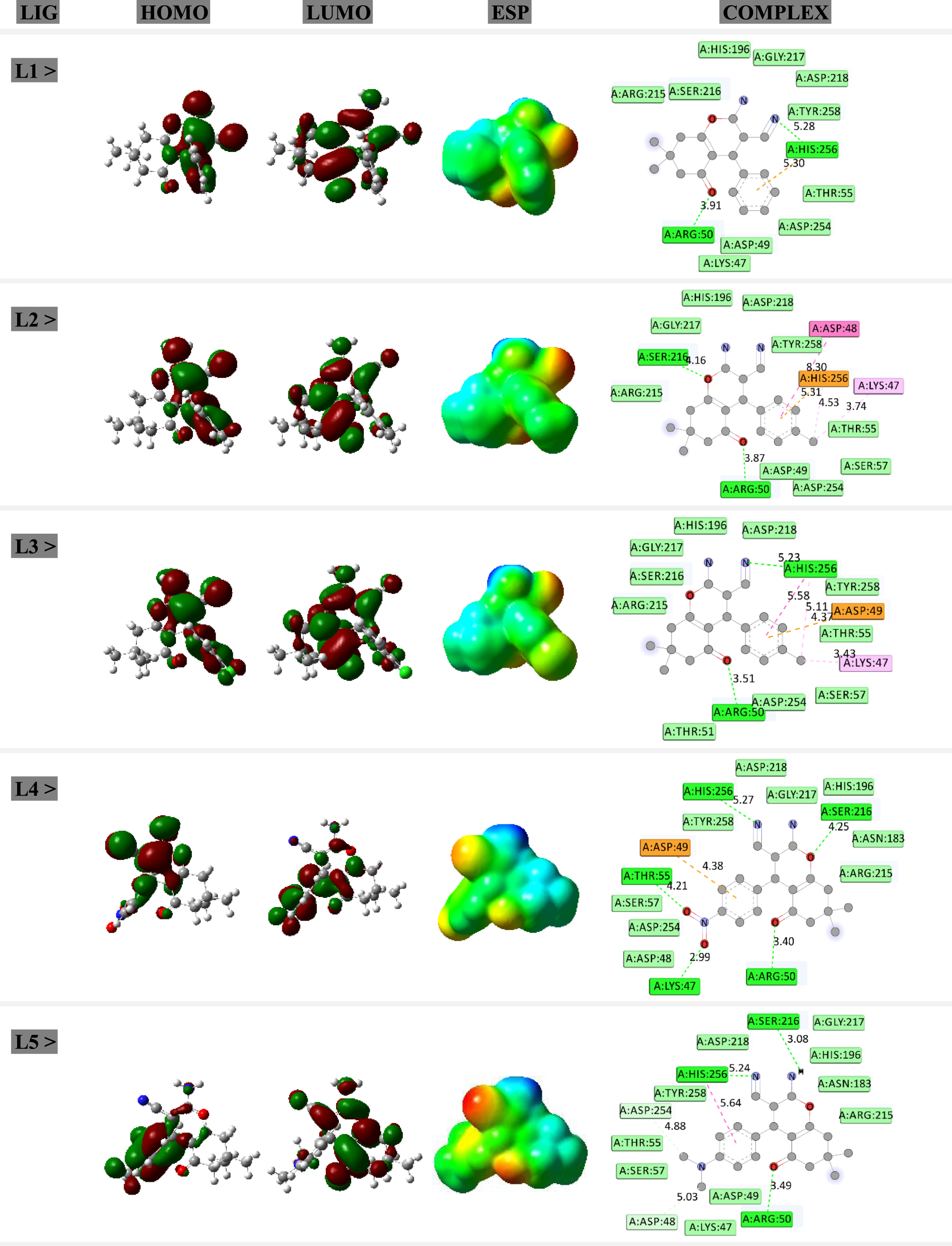
HOMO-LUMO distribution patterns, ESP surfaces, and ligand-target complexes. The green-red colors of HOMO-LUMO imply for positive-negative charges. The blue-light blue-green-yellow-red colors of ESP imply for positive-slightly positive-neutral-slightly negative-negative charges. The green-light green-orange-violet colors of complex imply for hydrogen bond-van der Waals-pi interaction-other noncovalent interactions of ligand-target models.
Within current work, five structures of chromene derivatives have been investigated for the purpose of electronic and structural analysis in addition to evaluating anticancer activity. As indicated in Table 1, the main skeleton of chromene derivatives were remained unchanged whereas the R group was changes according to building other model structures. It is important to mention here that one of procedures for drug design processes is lead optimization, in which one structure with unique skeleton will be modified by addition of functional groups [29]. To obtain the minimized energy structures, all five compounds were fully optimized to prepare the ligand structures for this work. To perform optimization calculations, the B3LYP exchange-correlation functional and the 6–31 + G* standard basis set were used as implemented in the Gaussian program [30]. To affirm reaching the global minimization, frequency calculations were performed avoiding the existence of any imaginary frequency for the optimized structures. As a consequence, the models were prepared and the evaluated quantum descriptors were listed in Table 1. The investigated ligand structures were designated by description of R group, molecular formula (MF) and molecular weight (MW). Furthermore, energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) were obtained for the optimized models in addition to evaluating energy gap (EG) difference between two levels. It is worth to note that the energy levels of each of HOMO and LUMO could indicate the electron transferring situation of a molecular systems, in which the HOMO level is designated for participating in electron donating process and the LUMO level is designated for participating in electron accepting process. Therefore, knowing such electronic features of molecular orbitals could help to show tendency of a molecule for contributing to electron transferring processes. Additionally, graphical representations of HOMO and LUMO distribution patterns and electrostatic potential (ESP) surfaces (Fig. 1) could help to predict molecular sites for participating in interactions with other substances. The colors of Red-Yellow-Green-Light Blue-Blue represent negative to positive charges with neutral charge of Green color in the middle. As a consequence, the ligand structures of chromene derivatives were prepared for participating in interactions with the cancerous enzyme target.
Molecular docking (MD) simulations were performed to obtain formations of ligand-target complexes (Fig. 1) using the AutoDock program [31]. The 3D macromolecular structure of methyltransferase enzyme (Code: 1hw4) was obtained from the Protein Data Bank [32]. It is important to mention that methyltransferase is an important enzyme involved in several types of cancers, in which inhibiting its activity could help to prevent cancer growth [33]. For doing MD simulations, the investigated ligand and target structures were interacted to each other in a grid box of 40*40*40 with 50 numbers of genetic algorithm conformational search. The obtained results of binding energy (EB) and inhibition constant (KI) were summarized in Table 2 in addition to graphical representations of interacting ligand-target complexes in Fig. 1.
Results and discussion
As described in Table 1, five structures of chromene derivatives have been investigated in this work for analyzing their electronic and structural features in addition to examining their anticancer activity. As shown by the representative skeleton of chromene, additional amino, cyanide, keto, and methyl groups are already available for the representative structure in addition to the R group, which differentiate the models from each other. The R groups are mainly phenyl based groups improving the surface are of the molecule for participating in interaction systems. As a consequence, five models were optimized to prepare ligand structures for investigating chemical and biochemical features of chromene derivatives. It is important to mention that existence of such chemical structures were already reported [34], in which their features and activity are investigated here to provide more insightful information about their applications by employing benefits of computational-based approaches for solving the problems [35, 36].
Based on performing optimization processes, structural geometries of five models of chromene derivatives were obtained. In this regard, the models were variated by the additional R group in the condition of remaining all other structural counterparts unchanged. As described in Table 1, the models were in suitable values of MW regarding the purposes of drug design processes to restrict such feature below 500 for the pharmaceutical compounds. Other features of R group and MF could show the structural components of investigated chromene derivatives. Analyzing energy values of HOMO and LUMO levels could show significance of addition of R groups to the original skeleton, in which the features of L1–L3 are almost similar to each other but they are different from those of L4 and L5. In L1–L3, both HOMO and LUMO levels detected the effects of additional R group; however, energy gap differences of two levels (EG) were remained almost similar. Comparing the additional R groups of these three models could show that the group is indeed a pure phenyl group with slight changes of para position with a methyl or chlorine group. However, that para position for phenyl of the R group was significantly changed for L4 and L5 compounds. Accordingly, HOMO and LUMO levels and their energy gap differences detected significant changes of the existence of additional R group. For L4, the levels of HOMO and LUMO were typically more achievable for electron transferring in comparison with those of other models. In other words, both of HOMO and LUMO levels went to more negative energies indicating moving to lower levels for catching the suitable position of electron transferring. Herein, in could be expected that L4 should contribute to stronger interactions with other substances. Indeed, such electronic molecular orbital features are very important for analyzing contribution of a molecule to electron transferring processes. Lower levels of HOMO and LUMO could mean that the values of energy barriers for electron transferring among interactions are reduces. In this regard, both levels could be considered as available levels of such electron transferrin processes. Moreover, values of EG are also very important for designating the internal electron transferring between HOMO and LUMO levels of a molecule, in which shorter distances could reveal easier electron transition inside the molecule. For this case, L5 was seen to be suitable for internal electron transition between HOMO and LUMO levels in comparison with other L1–L4 structures. However, levels of HOMO and LUMO are less suitable for participating in interactions with other substances in comparison with other L1–L4 structures.
As a consequence, the models were predicted regarding their electronic and structural features for how to participate in future interactions with other substances. Indeed, such features could be achieved by preforming computer-based works to recognize the nature of chemical structures prior to analyzing their biological activity. Accordingly, Fig. 1 exhibits that the analysis of HOMO and LUMO distribution patterns and ESP surfaces could reveal insightful information about molecular sites of structures regarding the electronic features. Localizations of the frontier molecular orbitals were designated by the distribution patterns and overall positive/negative charges were designated by the ESP surfaces. Interestingly, analysis of representations of ligand-target complexes could show that the main interactions of L1–L5 ligands with the enzyme target were from those molecular sites with localized HOMO distribution. This is indeed an important achievement in order to show significant roles of molecular sites of a structure for participating in interactions with other substances. In this case, the models were analyzed regarding their frontier molecular orbital features.
Further analysis of the results were for formations of interacting ligand-target complexes to show anticancer activity of the investigated chromene derivatives. To this aim, formations of interacting complexes were examined for each of L1–L5 chromene derivatives and methyltransferase enzyme. As mentioned earlier, overexpression of methyltransferase enzyme could lead to cancer growth, in which investigating efficient inhibitors for preventing its activity is a must. Therefore, such hypothesis of methyltransferase inhibition by assistance of chromene derivatives were examined here in this work. As shown in Fig. 1, five interacting ligand-target complexes were obtained by performing MD simulations. The graphical representations of complexes could show that the models were different in types and numbers of involving interactions between the central ligand and surrounding amino acids of target. To this point, different values of binding energy (EB) and inhibition constant (KI) could be expected for the investigated complex systems. As listed in Table 2, the strength of obtained complexes could be arranged in this order: L4 > L3 > L2 > L1 > L5. It could be remembered here from last part about analyzing the ligand structures, in which it was expected that L4 would participate in the strongest interactions and L5 would participate in the weakest interactions among the L1–L5 molecular ligand models. Interestingly, such predictions were affirmed by analyzing the strength level of obtained complexes. Indeed, this is an advantage of performing systematic computer-based works to solve problems regarding the electronic and structural features of ligands and predicting their future activity. As a consequence, the models were recognized based on their anticancer activity, in which all L1–L5 could interact with the methyltransferase enzyme to make stable complexes. Moreover, L4 was significantly seen the best ligand for playing this role by participating in the strongest interactions with the target among L1–L5 ligand models. The smallest value of KI for L4 among all investigated complexes could also reveal meaningful ignition of this ligand against the cancerous enzyme target. Details of such interacting systems could be recognized by graphical representations of Fig. 1 for interacting ligand-target complexes. The classical hydrogen bond interactions and other pi interactions were shown by dashed lines whereas those of van der Waals interactions were shown only by locating amino acids around the centrals ligand. It could be found that van der Waals interactions have higher contributions to interactions in comparison with other interaction types. As a consequence, types of amino acids and interactions were shown for knowing details of interactions between the central ligand and surrounding amino acids of the methyltransferase enzyme target. Comparing the results of this work with those of another earlier work on inhibiting the activity of methyltransferase enzyme [33] could show that the investigated structures of this work could be considered as useful ligands for participating in efficient interaction with the target enzyme. As a result, this work could provide information about the methyltransferase enzyme activity inhibition.
Conclusion
Within this work, the major goal of exploring some of representative chromene derivatives with efficient anticancer activities was investigated. To achieve this goal, combinations of ligand analyses and interacting with targets were used. In this regard, five models of chromene derivatives were investigated based on similarity of the original skeleton and addition of R group to make the models. The stabilized structures were obtained by performing DFT calculations. Electronic features were evaluated, in which the results indicated significant effects of existence of additional R group on the energy level of HOMO and LUMO of the models. By analyzing such characteristic frontier molecular orbital features, the models were arranged regarding their predicted role for participating in interactions, introducing L4 as the suitable ligand. Further investigations by preforming MD simulations for examining interactions of each ligand structure and methyltransferase enzyme target affirmed the already predicted results, in which L4-target was the strongest interacting ligand-target complex. By concluding such results, the investigated chromene derivatives were seen suitable for inhibiting activity of methyltransferase cancerous enzyme target to play anticancer role, in which their affinity for formations of interacting ligand-target complexes were shown by assistance of quantities of energies and qualities of representing the molecular counterparts and features.