
Abstract
In this work, we synthesized some organosilicon derivatives of piroxicam. Due to some properties of organosilicon compounds, including increased lipophilicity and thermal stabilization and prodrug for drugs, some silyl ethers of this drug were synthesized and characterized. Increasing of the lipophilic properties of this drug can be very important in the rate of absorption and its effectiveness. We synthesized silyl ethers of this drug, in the next step some copolymers this drug was synthesized. All of compounds were identified by spectroscopic methods.
Introduction
Piroxicam is a painkiller and its main use is to reduce or stop pain. In osteoarthritis, this drug has anti-inflammatory effects. This drug is used to treat many diseases such as headache and toothache, leg pain and piroxicam reduces the production of prostaglandins by controlling cyclooxygenase, thus showing its effectiveness in reducing and eliminating pain. It is also used to relieve joint, bone and muscle pain. It is even used to control gout and menstrual cramps. It binds to a large amount of protein and is metabolized in the liver and then excreted in the urine. For long-term storage of piroxicam, it is recommended that it be stored at –20°C, which should be stable for at least two years [1, 2].
In recent years, after 2001, the application of silicon in biotechnology has received much attention and many studies on the development of biocatalytic products from silicon polymers, release systems for enzymes, strong biosynthetic systems and Enzyme have been formed in the formation of silicon-carbon bonds [3]. Carbohydrates are one of the most numerous compounds in the biosphere that play a major role in cellulose synthesis. The use of silicon and silicon-based protecting groups in the synthesis of carbohydrates was first proposed in 1970 [4–6].
Silyl ethers are among the most widely used protecting groups for the alcohol functionally because the rate of deprotection can be modulated by simply altering the substituents on the silicon atom. As a result, the synthesis of small-molecule silyl ether has been explored using a variety of acid sensitive silane attachments including trimethyl silyl ether (TMS), triethyl silyl ether (TES), and triisopropyl silyl ether (TIPS). Three types of silyl ether prodrugs a) small molecule monofunctional silyl ether, b) polymeric monofunctional silyl ether prodrug, and c) polymeric asymmetric bifunctional silyl ether prodrug was used [7–9].
Piroxicam (Fig. 1) is a phenolic drug. These drugs are difficult to pass through the lipid barrier, and the presence of lipophilic groups, such as organosilicon, will increase this property, that is, increase the property of the drug, so the introduction of these groups will modify the property of the drug. For this reason, we did this work with various silyl groups.

Piroxicam structure.
Materials & apparatus measurements
Fourier transform infrared spectroscopy (FT-IR) analysis were recorded by a Bruker Vector-22 FT-IR spectrometer. The melting points were measured on an Electrothermal Melting Point Apparatus Model 9100-2. Proton nuclear magnetic resonance (1H-NMR) were recorded by a Bruker Spectrospin Avance 400 MHZ. Thermogravimetric analysis (TGA) of samples was determined by Mettler Toledo, TGA/SDTA851 e. All starting materials and reactants were commercially available and were purchased from Merck. The commercial reagents were used without prior repurification. All reactions were done in the argon atmosphere.
Synthesis of piroxicam silyl derivatives
Synthesis of triethylsiloxypiroxicam
First, 1.5 mmol (0.5 g) of piroxicam, 0.2 ml of trimethylchlorosilane and 0.30 ml of triethylamine as base in 20 ml of dry THF were mixed for 40 hours under argon atmosphere, then it was stirred at ambient temperature with a stirrer and the progress of the reaction was monitored by TLC. After 40 hours the product was subjected to thin layer chromatography using N–Hexane and ethyl acetate system with ratios of 7 : 3. The product was evaporated under vacuum to obtain a pure yellow solid.
Synthesis of dimethyl-tert-Bu-siloxypiroxicam
The process is similar to the previous, the time, after 45 hours the product was subjected to thin layer chromatography using N–Hexane and ethyl acetate system with ratios of 7 : 1. The product was evaporated under vacuum to obtain a pure yellow solid.
Synthesis of dimethylvinylsiloxypiroxicam
The process is similar to the previous. After 45 hours the product was subjected to thin layer chromatography using N–Hexane and ethyl acetate system with ratios of 7 : 1. The product was evaporated under vacuum to obtain a pure yellow solid.
Synthesis of copolymers
Synthesis of co-poly-ethylene-dimethylvinylsiloxypiroxicam with Methacrylic acid (MAA)
10 mg (0.024 mmol) of resulting vinyl dimethyl siloxypiroxicam monomer 0.56 ml of methacrylic acid (ratio of 1 : 3), 0.03 g of AIBN radical initiator and 30 ml of dry THF as solvent were mixed in round bottom flask, and was heated in 70°C for 72h. After cooling the product and washing with cooled methanol for three times, the resulting was evaporated to obtained white product. This reaction was repeated with ratio of 1 : 5 of monomers.
Synthesis of co-poly-ethylene-dimethylvinylsiloxypiroxicam with Syrene
10 mg (0.024 mmol) of resulting vinyl dimethyl siloxypiroxicam monomer 0.072 ml of styrene (ratio of 1 : 3), 0.03 g of AIBN radical initiator and 30 ml of dry THF as solvent were mixed in round bottom flask, and was heated in 70°C for 24 h. After cooling the product and washing with cooled methanol for three times, the resulting was evaporated to obtained white product. This reaction was repeated with ratio of 1 : 5 of monomers.
Results and discussions
As mentioned in previous section piroxicam is one of the important drugs, but this drug is one of compounds have hydrophilic property. The enhanced lipophilicity afforded to these compounds by the addition of silicon containing groups is expected to improve some properties. Increasing the lipophilicity of the drug causes the drug to pass quickly through the lipid barrier, thus multiplying the properties and effects of the drug. Cleavage of the silicon-drug bond is affected by chemical hydrolysis, a process less prone to interpatient variability than the metabolic process required for activation of conventional prod rugs. [10]. In other works, neurotropic and antitumor activities of pyridyl-substituted 5-Si-soxazolines have been investigated, and these compounds have more potency in protection against hypoxia than their analogues [11]. Also, silatranes are intracomplex organosilicon tris(2-hydroxy-alkyl) amine esters that have exhibited a broad spectrum of biological activity 12 –14]. In addition to these compounds other organosilicion compounds were synthesized and their biological activities were studied [15, 16]. Because of this, we synthesize some silyl ethers of piroxicam, and some network polymers of it. Incorporation of silyl ether into piroxicam not only increases its effectiveness but also causes a long-term effect as a prodrug.
Synthesis of piroxicam silyl derivatives
This derivative can be prepared by simple method, i.e. coupling chlorotrialkylsilanes in the presence of triethylamine as base, and in solvent such as THF, DMSO, and Deep Eutectic Solvent (DES) [17]. We used THF as solvent for our reactions. The performed reaction is shown in Scheme 1.

Reaction of organosilicon compounds with piroxicam.
Incorporation of silyl groups in all products were confirmed by FT- IR These groups in our silyl ethers (R3Si-) are: 1) R3 = Et3, 2) R = Me–R = Me–R = tert-Bu, 3) R = Me–R = Me–R = vinyl. Absorption in 1261 cm–1 is related to C-Si stretching vibrations, peak in 1090–1100 cm–1 is related to Si-O stretching vibrations, and in 820–830 cm–1 is related to C-Si stretching vibrations. These data conform the silyl groups in all of product. We, also showed the presence of piroxicam link in product. Absorptions of 3340 cm–1 and 3393 cm–1 are related to N–H stretching vibrations, adsorptions in 2958, 2918 cm–1 are related to aliphatic C–H stretching vibrations. Absorption in 1708 cm–1 is related to C = O stretching vibrations, peak in 1180 cm–1 is related to SO2 stretching vibration. These data also conform presence of piroxicam. In all products we can investigate and conform structures. Because all the FT-IRs of these materials are similar and also the changes in the peaks were similar, only one FT-IR is given below to prevent duplication (Fig. 2).
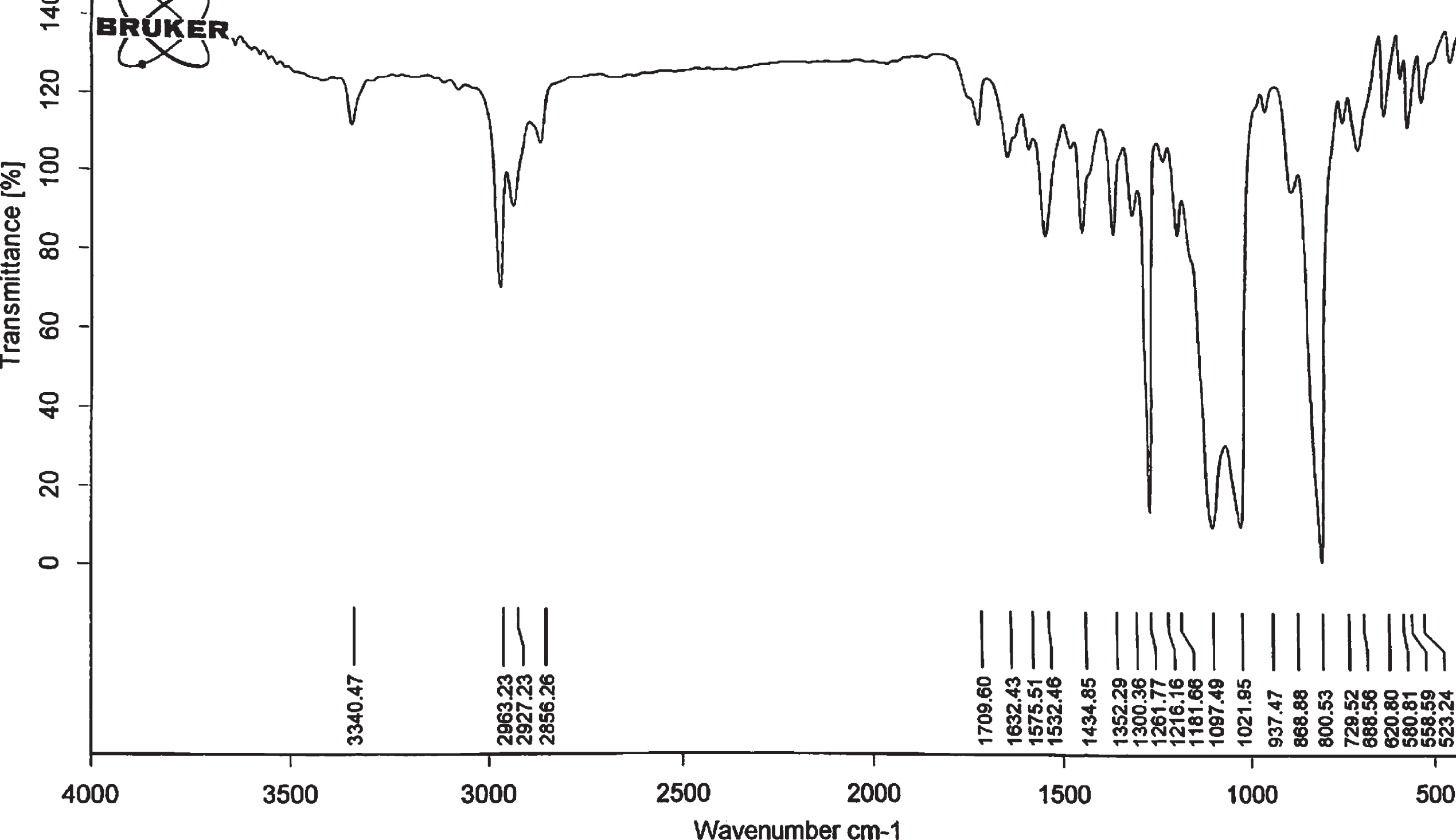
FT-IR spectrum of piroxicam silyl derivatives.
FT-IR (KBr) (ν/cm–1): 3340 (N–H stretch), 2856, 2927, 2963 (–C –H stretch), 1709 (C = O stretch), 1181 (SO2 bend), 1575 (C = C stretch), 1261 (Si–C stretch), 1097 (Si–O stretch), 800 (Si–C bend).
As shown in previous section, all groups in silyl ethers were investigated and approved. Also, all protons in piroxicam were identified by 1H-NMR.
3.1.2.1. 1H-NMR of triethylsiloxypiroxicam. The very strong and sharp peak of 2.85 ppm corresponds to 3 protons, carbon attached to nitrogen adjacent to SO2. Strong and multiple peaks of 7.2–8.4 ppm correspond to the 8 protons of aromatic rings. The very weak and wide peak around 12 ppm corresponds to the proton attached to the nitrogen adjacent to the carbonyl group. Relatively strong ternary and multiple peaks of 0.85–1.25 ppm are related to the external protons of the methyl group related to the attached silica. Relatively weak peaks of several 0.4–0.5 ppm correspond to the more internal protons of the ethyl group related to the binding of silyl (peak ratio is 1.5) Fig. 3.

1H-NMR spectrum of triethylsiloxypiroxicam.
1H-NMR (400MHz, DMSO) (δ/ppm): 2.85 (s, N - CH3, 3H), 7.2–8.4 (m, -C–H Benzen ring, 8H), 12 (s, N–H, 1H), 0.85–1.25 (t, -CH3, 9H), 0.4–0.5 (m, -CH2, 6H).
3.1.2.2. 1H-NMR of dimethyl-tert-Bu-siloxypiroxicam. A single branched peak at 0.82 ppm corresponds to 6 dimethyl protons of silicon, a peak at 2.2 ppm to 9 protons of the tert-butyl group that are not directly attached to silicon. The single-branch peak at 2.8 ppm corresponds to 3 carbon protons attached to adjacent nitrogen. Multiple peaks of 7.2–8.3 ppm correspond to 8 protons of aromatic rings. The very weak and wide peak around 12 ppm corresponds to the nitrogen attached to the adjacent carbonyl group (proton of amide) Fig. 4.

1H-NMR spectrum of dimethyl-tert-Bu-siloxypiroxicam.
1H-NMR (400MHz, DMSO) (δ/ppm): 2.85 (s, N - CH3, 3H), 7.2–8.4 (m, -C–H Benzen ring, 8H), 12 (s, N - H, 1H), 1.25 (s, t - Bu, 9H), 0.82 (s, (CH3) 2, 6H).
3.1.2.3. 1H-NMR of dimethylvinylsiloxypiroxicam. The peak at around 0.88 ppm corresponds to the 6 protons of the methyl group attached to silicon and the peak at the single-branch peak at 2.82 ppm corresponds to 3 carbon protons attached to adjacent nitrogen. The peak at about 5.5 to 6 ppm is for vinyl protons attached to silicon Multiple peaks of 7.2–8.3 ppm correspond to 8 protons of aromatic rings (Fig. 5).

1H-NMR spectrum of dimethylvinylsiloxypiroxicam.
1H-NMR (400MHz, DMSO) (δ/ppm): 2.85 (s, N - CH3, 3H), 7.2–8.4 (m, -C–H Benzen ring, 8H), 12 (s, N - H, 1H), 5–6 (H- vinyl, 3H), 0.82 (s, (CH3) 2, 6H).
TGA profile of piroxicam is shown in Fig. 6a. In the TGA profile of Me2VinylSiCl in Fig. 6b, weight loss of 70–320°C is related to evaporation of free water and weight loss of 320–500°C is related to the destruction of the weakest bond. The third weight loss, which occurs above 500°C, is related to the complete destruction of the compound. In the TGA profile of Et3SiCl in Fig. 6c, weight loss of 170–340°C is related to free water evaporation and weight loss of 340–470°C is related to the destruction of the weakest bond and the third weight loss, which occurred above 470°C, is related to the complete destruction of the compound. In the TGA profile of Me2t-BuSiCl in Fig. 6d, weight loss of 70–340°C is related to evaporation of free water and weight loss of 340–510°C is related to the destruction of the weakest bond. The third weight loss that occurs above 510°C is related to the complete destruction of the compound. The above profiles show that Et3SiCl is more stable than other celestial derivatives and begins to degrade later, and also degrades earlier than other derivatives or in other words at a relatively lower temperature than other derivatives.

TGA curve of a) piroxicam, b) Me2VinylSiCl, c) Et3SiCl, d) Me2t-BuSiCl, e) Me3SiCl.
Silyl ethers are among the most widely used protecting groups for the alcohol functionally because the rate of deprotection can be modulated by simply altering the substituents on the silicon atom. As a result, the synthesis of small-molecule silyl ether prodrugs has been explored using a variety of acid sensitive silane attachments including trimethyl silyl ether (TMS), triethyl silyl ether (TES), and triisopropyl silyl ether (TIPS). Although these materials are labile in vivo they are typically fastidious because of their vulnerability to acidic workups [18]. This limitation can be alleviated by incorporating silyl ether prodrugs within a polymeric drug delivery system. The combination of a small molecule drug with high molecular weight polymer provides protection for the therapeutic, and reduces the rate of degradation. Previously, polybutadiene and polyamine polymers have been functionalized with monofunctional (silyl ether prodrugs) of antiulcer prostaglandins [19, 20], which were designed to degrade under the harsh acidic environment found in the stomach.
Synthesis of copolymers of dimethylvinylsiloxypiroxicam with methacrylic acid (MAA)
In the next step of work, some network polymer of vinyl siloxy piroxicam derivatives were synthesized. Due to the fact that MAA has several applications in various fields, including in drug delivery systems, we prepared the copolymer of this monomer with vinyl siloxy piroxicam with ratio of 1 : 1, 1 : 3 and 1 : 5 in the presence of AIBN as radical catalyst (Scheme 2).

Copolymerization dimethylvinylsiloxypiroxicam with MAA.
3.2.1.1. FT-IR Spectroscopy. All of polymers were identified with FT-IR spectroscopy, peaks of functional groups of monomers, and MAA, also silyl groups are seen. This evidence confirms the existence of vinyl siloxy piroxicam MAA, and silyl group in network polymers. In the region 2400–3400 cm–1, the broad band is related to the stretching vibrations of the OH group related to methacrylate, which is broadened due to the presence of hydrogen bonds. In 1692 cm–1, a wide and strong peak is observed, which is related to the coupling of stretching vibrations of the carbonyl group in the polymer structure and also the carbonyl group in the synthesized compound building, and in the area 803 cm–1 peaks related to bending vibrations. Si-C bonding is observed as the peak of this series of spectra, which indicates the entry of dimethyl vinyl siloxypiroxicam into the polymerized compound (Fig. 7).

FT-IR of Poly vinyl siloxy piroxicam MAA.
3.2.1.2 TGA analysis. TGA of all polymers shows that increasing of thermal stability of piroxicam. This modification can be very important in this drug, i.e., increasing of thermal stability can be used in drug life and storage time. In the above TGA diagrams, three stages of degradation are quite visible, with a degradation of 200°C related to the available moisture. Degradation of polymer chains and hanging groups occurs in the range of 200–450°C and above 450°C is related to complete degradation of the compound (Fig. 8).

TGA curve of Poly vinyl siloxy piroxicam MAA with various ratio.
Vinyl dimethyl siloxypiroxicam and styrene monomer copolymers with 3 : 1 and 5 : 1 ratio were prepared under radical polymerization conditions. AIBN initiator (2,2-azo-bis-isobutyro nitrile) was used for copolymerization. The reaction time for polymerization was 24 hours and the reaction was performed without solvent. The resulting copolymers were crystalline and colorless. The polymerization conditions and the structural form of the resulting copolymers are shown in Scheme 3 and acetone and water were used as anti-solvents.

Copolymerization of vinyl siloxy piroxicam with styrene.
3.2.2.1. FT-IR Spectroscopy. In the area of 3400 cm–1, the broadened bond is related to the moisture in the composition, which is due to the presence of hydrogen bonds. Peak at 1703 cm–1, which is related to the coupling of stretching vibrations of the carbonyl group in the polymer structure and also the carbonyl group in the synthesized composite structure, and in area of 841 cm–1, the peak related to the bending vibrations of the Si-C bond as the prominent peak of this series of spectra. It can indicate the entry of dimethyl vinyl siloxypiroxicam into the polymerized compound (Fig. 9).

FT-IR of Poly vinyl siloxy piroxicam styrene.
3.2.2.2. TGA analysis. In the above TGA diagrams, three stages of destruction are quite visible, which include; below 200°C is related to the available humidity, degradation of polymer chains and hanging groups occurs in the range of 250–450°C and above 450°C is related to complete degradation of the compound (Fig. 10).

TGA curve of Poly vinyl siloxy piroxicam styrene with various ratio.
In this work, we synthesized silyl ether of piroxicam for improving of some properties of this drug. This work can be used in drug delivery system, such as improving thermal stability, storage time, and lipophilicity of phenolic drug, especially for piroxicam ointment. By increasing the lipophilicity of the drug, it easily passes through the lipid barrier and its effectiveness is significantly increased. From these points, this work can have important applications. We are working in some applications of it.
Footnotes
Acknowledgments
The authors gratefully acknowledge the research council of Azarbaijan Shahid Madani University for financial support.