
Abstract
The mechanism of cycloaddition reaction between phosphenium cation and phosphindene with formaldehyde has been systematically investigated at the B3LYP/6-311++G(d,p) level of theory in order to better understand the reactivity for the valence isoelectronic of carbene. Phosphenium cation acts as an electrophilic reagent and accepts σ electrons of formaldehyde to form a complex in the first addition step. The greater the positive charge on phosphorus atom in phosphenium cation, the more stable the formed complex is. Introduction of substituents will decrease positive charge on phosphorus atom in phosphenium cation. The order of positive charge on phosphorus atom is HP+–F > HP+–OH > HP+–NH2, which is consistent with their Lewis acidities. The complex transforms to a three-membered ring product via a transition state in the second cyclization step. The product is less stable than the complex due to its tension of small ring.
Introduction
Phosphenium cation (R2P+) is valence isoelectronic and isolobal with carbene (R2C), silylene (R2Si) and nitrenium cation (R2N+) [1, 2]. The simplest phosphenium cation is H2P+, which contains a positive charged phosphorus center and two hydrogens. H2P+ is detected through photoionization of PH3 and other phosphorus-bearing compounds [3]. The ground state of H2P+ is singlet, the energy gap of singlet-triplet is about 16 kcal/mol [4]. The hybridization of the central phosphorous atom can be regarded as being sp2, with a lone electron pair in the filled sp2 orbital and a vacant p orbital [5]. Introduction of a variety of substituents has been used to prepare and isolate stable phosphenium cations. Diaminomethylphosphenium cation, (NMe2)2P+, is the first stable, isolable cation in low-coordinated phosphorus chemistry [6]. Later, the other stable phosphenium cations have been characterized successively, for example the phosphanetriylammonium cation (MeN = P+) [7] and methylenephosphenium cation (Me2C = P+) [8].
The chemistry of different species tends to be dominated by their electrophilic behaviour, a key concern is their Lewis acidities. The fluoride ion affinities (FIAs) can act as a measure of the Lewis acidities of phosphenium cations [9]. Slattery et al. calculated the FIAs of 33 phosphenium cations with a range of substituents. The results demonstrated that phosphenium cations are often stronger Lewis acids than neutral species, but in many cases are less Lewis acidic than highly electrophilic cations such as [Me3C]+ or [Me3Si]+ [10]. The dipole moments of PH2+ and other compounds containing silicon or phosphorus have been computed to evaluate the intensities of rotational transitions and reactivity [11].
The chemical reactions of the phosphenium cation are similar with its carbon-based and silicon-based analogues, including cycloaddition reaction, C-H insertion reaction, etc., making them versatile reagents in kinds of transformations and reactions [12–14]. Phosphenium cation can be applied as ligands in coordination chemistry, acting as the building blocks in phosphorus-bearing compounds [15]. In addition, as a class of strongly π-accepting ligands, phosphenium cation can be used in homogeneous catalytic systems [16].
The reactions of carbene and silylene have been investigated extensively. The reactions of phosphenium cation have also attracted attentions [17, 18]. Since phosphenium cation have a vacant p orbital and a filled sp2 orbital, they can act as both Lewis acids and Lewis bases, respectively. Phosphenium cation can participate in complexation reactions with metals where it acts as a Lewis base [19, 20], or in cycloaddition reactions with alkynes where it acts as a Lewis acid [21]. In the present study, we performed comprehensive theoretical investigation of the reaction between phosphenium cation and formaldehyde. The reaction mechanism has been described and the effect of a variety of substituents in phosphenium cation has been evaluated. The present results will enrich the available data for the relevant phosphenium cation chemistry, and will provide more information for expanding its application in coordination chemistry and catalytic systems.
Calculation method
For the reaction between phosphenium cation and formaldehyde (HCHO), the popular hybrid density functional B3LYP method [22, 23] with 6–311++G(d,p) and AUG-cc-pVTZ basis sets have been employed to locate all the stationary points along the reaction pathway without imposing any symmetry constraints. The reliability and efficiency of this method in predicting the geometries and properties have been verified by a number of investigations [24, 25]. Frequency analyses have been conducted to confirm the nature of the minima and transition state. Moreover, intrinsic reaction coordinate (IRC) calculations have also been made to further validate the calculated transition state connecting reactant and product. Additionally, the relevant energy quantities, such as the reaction energies and barrier heights, have been corrected with zero-point vibrational energy (ZPVE) corrections.
To further refine the calculated energy parameters, single-point energy calculations have been performed at the CCSD(T)/6–311++G(d,p) and CCSD(T)/AUG-cc-pVTZ levels of theory based on the stationary points optimized at the corresponding B3LYP method. As described below in Fig. 1, Tables 1 2, several kinds of computational levels can give approximatively consistent results for the optimized structures and the calculated reaction profile of the reaction between H2P+ and HP with HCHO. Therefore, as for the reaction of substituted phosphenium cation (XYP+) and HCHO, B3LYP method in conjunction with the 6–311++G(d,p) basis set have been performed to confirm the nature of the minima and transition state as well as the energy parameters. For the sake of simplicity, for all the reactions, the energy results at the B3LYP/6–311++G(d,p) level have been mainly discussed below if not noted otherwise.

Optimized structures of the reactants, complex, transition state, and product in the cycloaddition reaction at the B3LYP/6–311++G(d,p) (first line) and B3LYP/AUG-cc-pVTZ (second line, italic) level of theory, where the bond length and bond angle are in angstrom and degree, respectively.
The calculated relative energies (in kJ/mol) in the reaction of phosphenium cation (H2P+) and formaldehyde with respect to the isolated reactants at the four calculated levels of theory considering the ZPVE corrections
The calculated relative energies (in kJ/mol) in the reaction of phosphindene (HP) and formaldehyde with respect to the isolated reactants at the four calculated levels of theory considering the ZPVE corrections
All the calculations have been performed using Gaussian 09 program [26].
For the cycloaddition reaction of phosphenium cation and formaldehyde, the first step is the formation of a complex (Com) without barrier. The second step is the Com isomerizes to a product (Pro) via a transition state (TS). The geometric parameters for the reactants (R1-H2P+ and R2-HCHO), Com, TS, and Pro involved in the cycloaddition reaction are displayed in Fig. 1. The corresponding energies are listed in Table 1.
When phosphenium cation approaches to the O atom of formaldehyde, it can form a complex (Com) with formaldehyde, which is a barrier-free process. In the Com, the configuration of formaldehyde fragment changed slightly compared with that in isolated formaldehyde. The bond length of C–O in the Com has been prolonged slightly by 0.033 Å than that in isolated formaldehyde, denoting the weakening of the C–O bond. To investigate the combined process of phosphenium cation and formaldehyde, the potential energy curve for the Com has been constructed along the distance between two fragments. As displayed in Fig. 2, the energy of the system decreases continuously before combination. After the equilibrium point (O-P bond is 1.876 Å), the energy will be increased rapidly as PH2+ and HCHO approaching to each other.
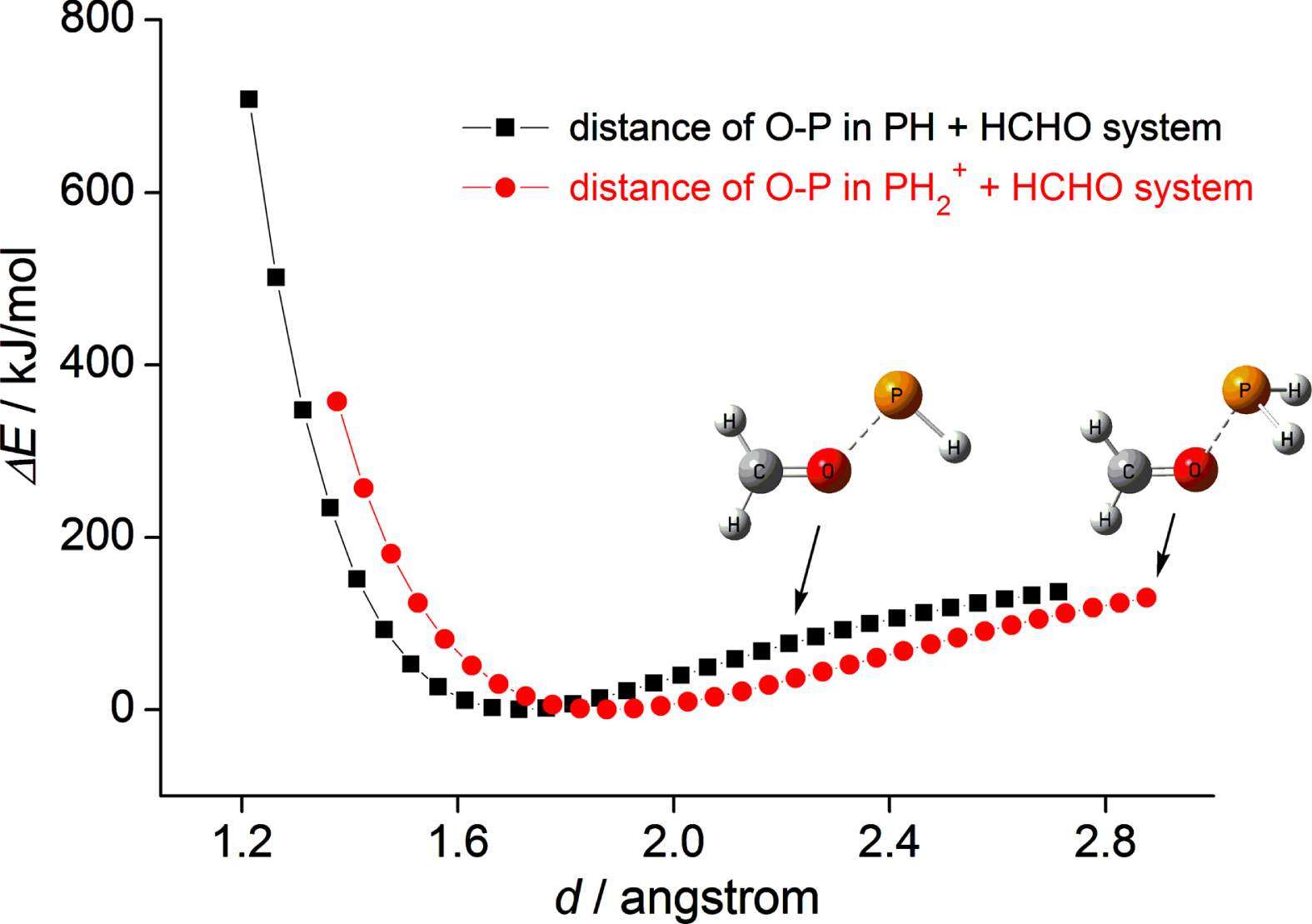
Energy changes in the combined process of complex along with the distance between two fragments at the B3LYP/6–311++G(d,p) level of theory.
The addition process can be understood through interaction of the frontier molecular orbital theory. As displayed in Fig. 3, the frontier orbitals for LUMO of H2P+ and HOMO of HCHO are symmetrical matching. As H2P+ initially interacts with HCHO, the 3p unoccupied orbital of phosphorus in H2P+ inserts into the sp2 orbital of HCHO to form a σ⟶p donor-acceptor bond, resulting in the formation of the Com.

The calculated LUMO orbital for H2P+ and HP, the HOMO orbital for formaldehyde.
The cycloaddition reaction of neutral phosphindene (HP) with HCHO, marked with superscript “HP”, is identical with that of H2P+. The geometric parameters for the reactant (HP) and stationary points (ComHP, TSHP, and ProHP) involved in the cycloaddition reaction are displayed in Fig. 1, the corresponding energies are listed in Table 2. Like as H2P+, the HP molecule has the 3p unoccupied orbital. The difference between HP and H2P+ is that the former has two pairs of lone electrons located in the two sp2 orbitals, the latter has only one pair of lone electrons in one sp2 orbital, the other sp2 orbital is unoccupied. To reduce repulsion between sp2 electrons of phosphorus in HP and σ electrons of C-H bond in HCHO, the distance of P and C in ComHP is elongated, which can be reflected with the angle of COP (133.2 in ComHP v.s. 123.5 in Com). In the process of combination of ComHP, as HP approaching to HCHO, the energy undergoes the minimum point (here the distance of O-P is 1.714 Å), which is similar to the Com (see Fig. 2).
H2P+ acts as an electrophilic reagent and accepts σ electrons of HCHO in the cycloaddition process. The greater the positive charge on phosphorus atom in H2P+, the easier the formation of the σ ⟶ p donor-acceptor bond will be, therefore, the more stable the formed complex will be. As displayed in Tables 1 2, the Com is exothermic with the value of 248.1 kJ/mol compared to the reactants. By contrast, the exothermic value of the ComHP is 150.8 kJ/mol. The reason is that H2P+ is a cation but HP is a neutral molecule. There is more positive charge on phosphorus atom in H2P+ than that in HP (Mulliken charge is 0.955 v.s. 0.084 for H2P+ and HP, respectively).
For the sake of better understand cycloaddition reaction of phosphenium cation and formaldehyde, the effect of variety of substituents has been considered. As displayed in Table 3, for the monosubstituted HXP+ and the disubstituted XYP+, the greater the positive charge on phosphorus atom in phosphenium cation, the more stable the formed complex will be. Take the monosubstituted HP+–OH as an example, both of phosphorus and oxygen are in sp2 hybridization. The unoccupied p orbital of phosphorus and occupied p orbital of oxygen are in parallel. The two p orbitals overlap to form a π bond, in which some electrons on oxygen transfer to phosphorus, resulting in decrease of positive charge of phosphorus in HP+–OH. The greater the electronegativity, the greater the ability of bounding electron is, therefore, the less the electrons transformed to phosphorus is. The electronegativity order of elements in the second period is F > O > N > C, therefore, the order of positive charge on phosphorus should be HP+-F > HP+–OH > HP+–NH2 > HP+-CH3, which is in good agreement with the calculated results with an exception of HP+-CH3. The reason is that the carbon in -CH3 is in sp3 hybridization, it can’t form π bond with the unoccupied p orbital of phosphorus, which limits the transfer of electron from C to P. The order of positive charge density in substituted phosphenium cation is consistent with the order of their Lewis acidities reported by Slattery’s group [10].
The calculated Mulliken charge of phosphorus atom in R1 and relative energies (in kJ/mol) in the reaction of substituted phosphenium cation and formaldehyde at the B3LYP/6–311++G(d,p) level of theory considering the ZPVE corrections
The Com transforms to the Pro via a cyclization process with a barrier of 114.4 kJ mol–1. The unique imaginary frequency calculated for the corresponding TS is 447i cm–1 at the B3LYP/6–311++G(d,p) level of theory.
As shown in Fig. 1, the distance of C-P in TS is 2.215 Å, indicating the new bond of C-P is to be formed. At the same time, the distance of C–O in R2 fragment of TS is reached to 1.290 Å, which elongated 0.055 Å than that in Com. Therefore, based on the bond length data, the double bond of C–O in Com is to be transformed into single bond in Pro via TS. The formation of new σ bond of C-P and the cleavage of π bond of C–O happened simultaneously. As shown in Fig. 4, those changes can be further validated by the IRC calculations on the basis of TS. The Pro is a three-membered ring compound, bearing phosphorus and oxygen, which is stabilized 189.9 kJ mol–1 compared to the reactants.
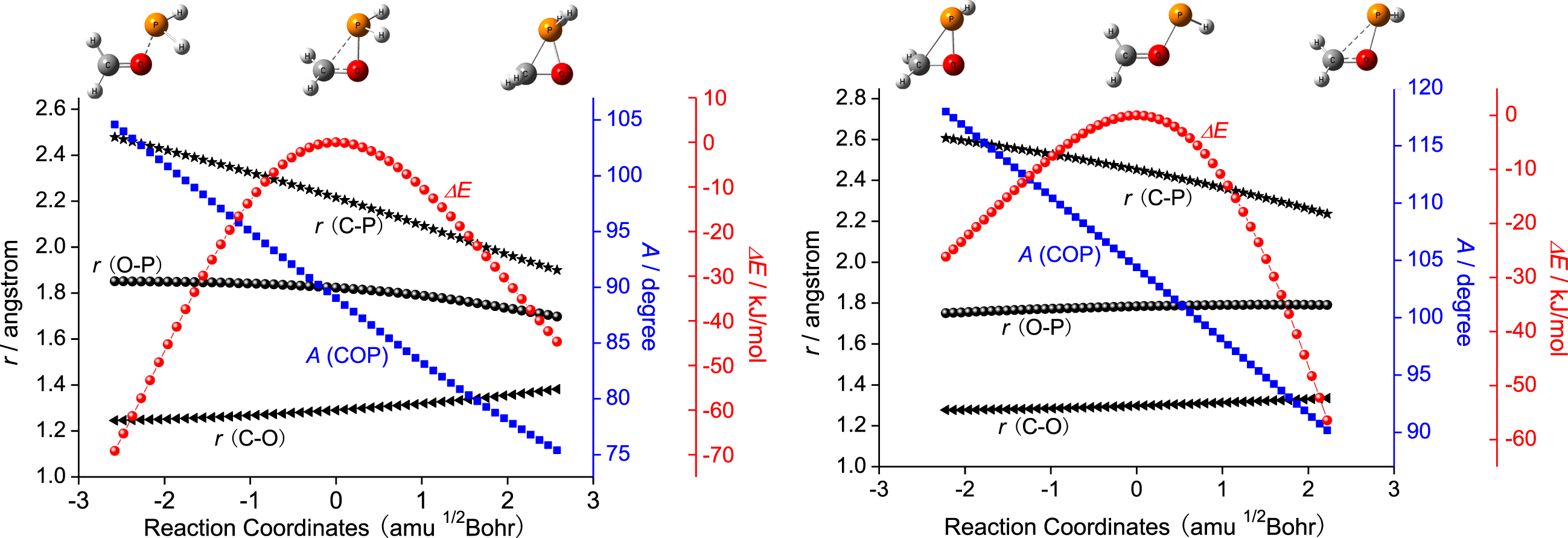
The relatively energy and geometric parameters changes along the reaction coordinates of the TS (up) and TSHP (bottom).
As displayed in Figs. 1 2, the cyclization process of ComHP is similar to Com. However, as summarized in Table 1, the barrier of the cyclization process of ComHP is much lower than that of Com (45.8 kJ mol–1 for the former v.s. 114.4 kJ mol–1 for the latter). Therefore, the cycloaddition reaction of phosphindene with formaldehyde is easier than that of phosphenium cation with formaldehyde.
In the present study, the cycloaddition reaction between phosphenium cation and formaldehyde has been comprehensively investigated employing B3LYP theoretical method with 6–311++G(d,p) and AUG-cc-pVTZ basis sets. The first step of the reaction is the formation of a complex without barrier. H2P+ acts as an electrophilic reagent and accepts σ electrons of formaldehyde in the addition process. The greater the positive charge on phosphorus atom in phosphenium cation, the more stable the formed complex is. Introduction of substituents will decrease positive charge on phosphorus in phosphenium cation. The order of positive charge of phosphorus is HP+–F > HP+–OH > HP+–NH2, which is consistent with their Lewis acidities. Therefore, introduction of substituents will decrease the stability of the complex. The complex transforms to a three-membered ring product via a transition state. The product is less stable than the complex due to its tension of small ring.
Footnotes
Acknowledgments
This work was supported by the Nature Science Foundation of Shandong Province (ZR2020MC004, ZR2015HL120), the Science and Technology Project of University of Jinan (XKY2030), the Project of Research Leader Studio of University Funding 20 items in Jinan (2019GXRC058), Technological SMEs’ innovation ability improvement project (2021TSGC1279), Clinical Science and Technology Innovation Program of Jinan (202019191), and Major Subject Projects of Key Disciplines of University of Jinan (1420707).