
Abstract
In this paper, we applied PC-SAFT and SAFT-HR equations of state so as to reproduce the solubility of carbon dioxide in aqueous diethanolamine solution. By using these equations, we have been able to model the solubility of carbon dioxide in aqueous amine solution in more than 350 experimental data points with wide range of amine molar concentration (0.01–0.12), temperature (300 K –478 K), carbon dioxide partial pressure (0.0001 KPa –5473 KPa), and carbon dioxide loading (0.04 –1.1). Ternary systems including water, carbon dioxide and diethanolamine have also been modeled by PC-SAFT and SAFT-HR equations of state based on bubble pressure algorithm. Binary interaction parameters are set to zero to show the genuine capability of equations of state in reproducing such experimental data. Provided modeling results have been obtained from MATLAB R2019b software for PC-SAFT equation of state are less deviated with experimental data. Overall average relative deviation of SAFT-HR and PC-SAFT are 45.452% and 4.374% respectively which show that PC-SAFT is a robust equation of state in predicting the solubility data of carbon dioxide in aqueous alkanolamine solutions.
Nomenclating
Helmholtz energy Coefficients of correlations Effective segment diameter Binary interaction parameter Mole based chemical equilibrium constant Liquid phase mole fraction, mole fraction in SAFT-HR EOS Vapor phase mole fraction Segment number Electronic charge (1.60219×10-19 C) Pressure Temperature Energy parameter Density Temperature-independent segment diameter Vacuum permittivity (8.85419×10-12 C2 J-1 m-1) Fugacity coefficients Association contribution Dispersion contribution Hard sphere contribution Electrostatic contribution Born contribution Born Residual Calculated properties Experimental properties Component i Binary pair of i and j Component j
Introduction
Carbon dioxide (CO2) is a colorless gas with acidic taste which is used in chemical processes such as methanol production as raw material, conditioning systems, plastics industry, food industry, oil industry and etc. CO2 is also known as greenhouse gas is able to trigger ozone depletion [1]. CO2 emissions from different sources like coal-fired power plants can be controlled by grabbing CO2 from emission resource and apply in in other units or industries. [2] At this time, the usage of chemical solvents especially aqueous alkanolamines for CO2 removal is the most advanced technology for the CO2 separation from flue gases. Alkanolamines can be scientifically classified into three main types which are known as primary, secondary and tertiary. Examples of these aqueous alkanolamines can be presented as monoethanolamine (MEA), diethanolamine (DEA), methyldiethanolamine (MDEA) and amino-methyl-propane (AMP). Primary and secondary type of alkanolamines such as DEA frequently exhibit high-rate CO2 absorption. However, tertiary alkanolamines such as MDEA show the opposite manner because MDEA has got low vapor pressure (1 Pa @ 20°C) and low evaporation loss of absorbent during CO2 capture. Although it is worth noting that DEA and MDEA are very applicable because they are less corrosive and consume less energy for regeneration [3, 4].
Thermodynamic modelling of the solubility of CO2 in aqueous alkanolamine is crucial in related chemical process designs such as CO2 capturing and sour gas sweeting. These kinds of aqueous solutions are vastly non-ideal and produce ionic species in chemical reactions. According to this fact, the requirement of sophisticated thermodynamic models which is capable to calculate vapor–liquid equilibrium (VLE) is comprehensively sensed. Two main approaches are generally applied for this modeling purpose [5]: γ - φ approach (the activity coefficient method): Excess Gibbs energy models is used for liquid phase calculations such as eNRTL model [6] and electrolyte UNIQUAC-NRF model [7]. φ - φ approach (The equation of state method): using Equation of State (EoS) for both vapor and liquid phases calculations such as Cubic Equation of State (CEoS) [8], electrolyte Cubic Square-Well (eCSW) EoS [9] and electrolyte Statistical Associating Fluid Theory (Huang and Radosz) (eSAFT-HR) EoS and Perturbed-Chain Statistical Associating Fluid Theory (PC-SAFT) EoS [10].
Perturbed chain statistical associating fluid theory (PC-SAFT) equation of state first was developed by Gross and Sadowski [11] based on statistical mechanical methods particularly, perturbation theory and spherical particles in the context of hard chains are utilized as reference fluid.
A large variety of experimental works and thermodynamic models have been presented due to predict the gas solubilities in different solutions [12–23]. For instance, Najafloo et al. have recently developed eSAFT-HR EoS in order to determine the nonelectrolyte part of the SAFT-HR EoS [10]. In another investigation done by Najafloo et al. the eSAFT-HR EoS has been successfully used in order to acquire activity coefficients of the species existed in strong electrolyte solutions in a wide range of concentration [24]. In addition, Najafloo et al. applied the eSAFT-HR EoS to model the solubility of CO2 in aqueous MDEA and 2-((2-aminoethyl) amino) ethanol (AEEA). According to this study the average absolute deviation percent (AAD %) of calculating the pressure as a function of CO2 loading was determined 7.74% at four different isotherms between 313.15 and 358.15 K [24]. Najafloo et al. have also applied SAFT EoS for modeling the solubility of CO2 in Monoethanolamine (MEA). The MEA pure properties, vapor-liquid equilibrium (VLE) properties of binary MEA-H2O and ternary MEA- H2O-CO2 are estimated by considering the SAFT modification of Huang and Radosz (SAFT-HR) [13]. Furthermore Najafloo et al. have studied the modeling of CO2 solubility in aqueous MDEA by applying successfully electrolyte type of SAFT-HR EoS in the wide ranges of temperature, pressure and concentration to determine the binary adjustable parameters. They have also determined the interaction parameters of ternary solution CO2–MDEA–H2O using 413 experimental data point based on bubble pressure algorithm [25]. Yazdi et al. have determined the solubility of hydrogen sulfide in the aqueous solution of methyldiethanolamine with PC-SAFT EoS, they have investigated a new method for calculating the association parameters [26].
In current study, the solubility of CO2 in aqueous alkanolamine solutions is considered by SAFT-HR and PC-SAFT equations of state, since the comparison of precision of SAFT-HR and PC-SAFT in three species systems, applicability of PC-SAFT EoS is investigated for the solubility of CO2 in aqueous MDEA+DEA solutions and the influence of perturbed chain terms on the equilibria is also considered. In this work, CO2 solubility in aqueous MDEA+DEA solutions is calculated without considering binary interaction parameters in the CO2+(MDEA+DEA+H(2O) system which includes four volatile components (CO2, H2O, DEA and MDEA).
Thermodynamic framework
SAFT-HR EoS
The residual Helmholtz free energy of the SAFT-HR EoS includes four terms shown as follows:
T, K, N, as well as A are represented as absolute temperature in Kelvin, Boltzmann’s constant, Avogadro’s number and Helmholtz free energy respectively. Superscripts res, hs, disp, chain and assoc are known as residual, hard sphere, dispersion, chain and association.
All of the terms of the equation (1) will be obtained from the following equation:
x, d, m, ρ,
K
ij
and v0 are the binary interaction parameter and are the temperature-dependent segment volume, respectively. D
ij
universal constants are accessible in reference [27].
κ A i B j , ɛ A i B j , Δ A i B j and X A i are volume of association, the energy of association, the association strength and mole fraction of molecules i not bonded at site A, respectively.
The radial distribution function g
ij
(d
ij
) is shown by:
In terms of molar Helmholtz free energy, PC-SAFT is expressed by:
Where ares is the residual Helmholtz free energy at the same temperature and density of the fluid of interest:
It is supposed that, in aqueous ethanolamine solutions, hydrogen bonding (association term, aassoc) dominantly contributes to the Helmholtz free energy of the solution and the polar contribution is less effective. The other contributions, however, cannot be ignored [27].
The hard-chain contribution to the Helmholtz free energy ahc accounts for the excluded volume in the fluid and the covalent bonds in the chain molecules:
We use the expression suggested by Boublik and Mansoori et al. [28, 29] for the excluded volume (ahs). The chain contribution to the Helmholtz free energy achain was derived by replacing association bonds with covalent bonds. Several attempts were made to improve the accuracy of the chain contribution by including the dimer effect on the chain formation. Because these modifications were not evaluated strictly, we decided to use the conventional expression suggested by Chapman et al. [30] Therefore, we get:
Where giihs(dii) is the hard-sphere radial distribution function at contact:
The two other parameters are defined by:
Where ρ is the molar density, m
i
is the number of segments in a molecule of component i, and NAv is Avogadro’s constant. The parameter dii describes soft repulsions between molecules. In terms of the segmental diameter σii and the depth of the square-well potential ɛii, the parameter dii is expressed by:
The dispersion contribution to the Helmholtz free energy adisp accounts for van der Waals forces. In earlier versions of the SAFT approach, the dispersion term was usually defined as a perturbation to the hard-sphere repulsive term (ahs). Gross and Sadowski [31] suggested a perturbation to the hard-chain repulsive term (m.a
hs
+a
chain
). The expression derived by Gross and Sadowski, is slightly complicated. However, comparisons made in predicting the VLE of normal and associating fluids using PC-SAFT and the SAFT version of Huang and Radosz [32, 33], revealed that PC-SAFT is more accurate than the SAFT version of Huang and Radosz. In this work we use the dispersion expression defined by Gross and Sadowski [31]:
Where Equation (22) is the compressibility of the hard-chain fluid:
In Equation (30), the mixing rules are defined by:
Where the conventional combining rules for ɛ
ij
and σ
ij
are employed:
In Equation (38), k ij is the binary interaction parameter that is zero for MDEA + water solution. For evaluating the VLE of normal fluids, inclusion of ahc and adisp in the PC-SAFT approach is sufficient. Three parameters, i.e., the number of segments (m), the depth of the square-well potential (ɛ/k), and the segment diameter (σ) are required to characterize each compound.
Helmholtz’s free energy of association is calculated by the relationship provided by Chapman, Gubbins, and Radosz [30]:
Where XAi is the mole fraction of molecule i that not participate in bounding at site A, and Mi is the number of sites on molecule i. The parameter XAi is given by Huang and Radosz [33]:
The right sigma is on all sites of the molecule j, and the sigma to the left represents the sum of all the components. In addition, ρj is the molar density of component j, and is expressed as:
Where ρ is the molar density of the solution. The association strength ΔAiBj is given by:
In Equation (44), if one sets i equal to j, the expression for gii(dii)hs given in Equation (25) will be recovered. The association energy ɛAiBj and the effective association volume κAiBj between associating substances i and j are determined from the association energies ɛAiBi and ɛAjBj and the effective association volumes κAiBi and κAjBj, respectively. The combining rules suggested by Wolbach and Sandler [34] are used. These combining rules were applied successfully by several investigators, e.g., Gross and Sadowski [19], Karakatsani et al. [35], and Kleiner and Sadowski [36, 37].These combining rules are:
Hence, if one of the substances in a mixture is non-associating, κAiBj and consequently ΔAiBj will vanish, i.e., no induced association will be considered.
In order to investigate the absorption of CO2 in aqueous alkanolamine solutions, we must simultaneously study the phase and the chemical equilibrium Reactions.
The chemical reaction within the liquid phase of CO2, DEA, MDEA, and H2O are as following:
The coefficient of activity is considered to be 1 in the calculation. K j , γ i and νi,j are the mole fraction based chemical equilibrium constant for the reaction j, the mole fraction, the activity coefficient (mole fraction scale) and the reaction stoichiometry of species i in reaction j, respectively. The values of K1, K2, K3, K4, K5 and K6 are presented in Table 1. [27]
Temperature-dependent equilibrium constants (based on mole fraction) a for reactions [27]
ln (Ki)=A+B/T+C ln(T)+DT.
Equations of electro neutrality and mass balance are considered as following:
In order to obtain the equilibrium concentration of species in the liquid phase, the mole balance and electro neutrality must be solved simultaneously in the Smith and Maison method [38–41].
Because of the low vapor pressure of MDEA and DEA at the temperature range of 300-473 K, their presence in the vapor phase is negligible. As a result, only CO2 and H2O molecules are present in the vapor phase. All ionic species are considered only in the liquid phase. The solution method is performed in the form of φ - φ approach. The equilibrium relation of the phases is as following:
In the above equation, x i and y i are mole fractions of species i in the liquid phase and vapor phase respectively. Superscript l and v refer to liquid phase and vapor phase respectively. The fugacity coefficients are calculated by eSAFT-HR and PC-SAFT EoS [42, 43].
Database
A large number of the CO2 solubility data in aqueous DEA solutions are available in the literatures. At this work, following work of Baygi and Pahlavanzadeh [44], we use a data set containing 355 ternary systems in the temperature range of 303–473 K and the CO2 loadings of 0.04–1.1 as listed in Table 2.
Table 2 shows the range of temperature and CO2 loading as well as number of extracted data points of experimental VLE data of CO2 absorption in aqueous DEA solutions which all are categorized by the name of the authors.
The pure component parameters of eSAFT-HR EoS are similar to the parameters of SAFT-HR EoS. The term born the adjustable parameters for eSAFT-HR EoS are segment number (m), temperature-independent segment volume (
The parameters of eSAFT-HR and PC-SAFT EoS for pure DEA are determined by simultaneous optimization of the saturated vapor pressure and the liquid density given by the following objective function:
In this work, water and carbon dioxide are considered as Bond–Band–Barrier (3B) correlation substance and non-correlated substance respectively [31]. Table 4, on the other side, presents water and carbon dioxide parameters which are extracted from literature [31]. According to the researches done by Zoghi et al, 3B and 2B correlations has been considered for MDEA and DEA respectively. In order to derive the most optimized SAFT-HR parameters by Equation 53 for MDEA and DEA, having vapor pressure and liquid density is necessary.
Pure component parameters of PC-SAFT EoS [27]
In the PC-SAFT equation of state k ij is the binary interaction parameter that is zero for MDEA+DEA+water solution. For evaluating the VLE of normal fluids, inclusion of ahc and adisp in the PC-SAFT approach is sufficient. Three parameters, i.e., the number of segments (m), the depth of the square-well potential (ɛ/k), and the segment diameter (σ) are required to characterize each compound (Table 4). However, in ethanolamine solutions, hydrogen bonding contributes dominantly to the nonideality of the ethanolamine solutions and its contribution should be considered.
In this work, for showing the predictability of the eSAFT-HR EoS, the binary interaction parameters (kij) are assumed to be zero for all of the binary subsystems.
Modeling MEA-CO2-H2O ternary system
Based on the previous studies done by researchers, binary interaction parameters (kij) are supposed to be tuned for the total number of binary subsystems so as to insure high predictableness level of the eSAFT-HR EoS. According to the chemical equations 52, the four molecular (MDEA, CO2, MEA and H2O) and seven ionic (H3O+, OH–, Chemical equilibrium, charge and mass balance equations are solved by Dawodu and Meisen [45] in the inner loop. The VLE calculation is computed by φ - φ approach in the outer loop.
The binary interaction parameters are determined by correlating the modeling results with experimental VLE data for the binary systems in Tables 3 and 4. Table 2 includes the ranges of temperature, DEA mole fraction, MDEA mole fraction, experimental pressure and loading. This paper also contains 350 isotherms along with data points that have all been extracted from literature and mentioned for each dedicated isotherm. Average Absolute Relative Deviation (AARD %) have also calculated by Equation 59 and reported in Table 5 6. According to the information in Tables 5 6, the overall AAD% is 45.542 for SAFT-HR EoS and 4.374 for PC-SAFT EoS, which represents an acceptable result for a ternary weak electrolyte solution.
Pure component parameters of SAFT-HR EoS [32]
Pure component parameters of SAFT-HR EoS [32]
bAverage absolue relative percent deviation (AAD%) =
The purpose of this research is to apply PC-SAFT and SAFT-HR for modeling the solubility of carbon dioxide in the in aqueous DEA solution. The fulfillment of this modeling task requires some steps to be accurately followed: Gathering experimental data from literature. Modeling the solubility of CO2 in aqueous alkanolamine solution by applying SAFT-HR and PC-SAFT equations of State.
In this work, we applied SAFT-HR and PC-SAFT equations of state for modeling the solubility of 350 CO2 / DEA experimental data points in wide variety of amine mole fraction (0.01-0.14), temperature (300-478 K), CO2 partial pressure (0.1-5500 kPa), and CO2 loading range (0.04-1.1). The calculations of ternary solution including CO2-H2O-DEA was carried out by using bubble point equilibrium algorithm in the reaction solution of both thermodynamic models. The binary interaction parameter (kij) was considered zero to show the predictive power of both models.
According to the data provided in this paper, we received the following outcomes: The model provided for PC SAFT is able to make satisfying predictions on the extracted experimental data. PC-SAFT and SAFT-HR are able to make better prediction in 413 K. An extensive examination in the result of modeling reveals that the AARD% is being reported higher than the expected range in some points with lower CO2 loading owing to the instrumental error. The absolute value of relative deviation is reported 4.374 % with PC-SAFT model, whereas this number is 45.54 % with SAFT-HR model. PC-SAFT makes superior predictions in low CO2 loading magnitude and pressure.

Comparison of calculated and experimental data of total pressure as a function of CO2 loading using SAFT-HR (dashed line) and PC-SAFT (Straight line), (at 303, 313, 323 and 338 K and DEA mole fractions (0.043); lines and points represent the model results and experimental data respectively [46, 49].
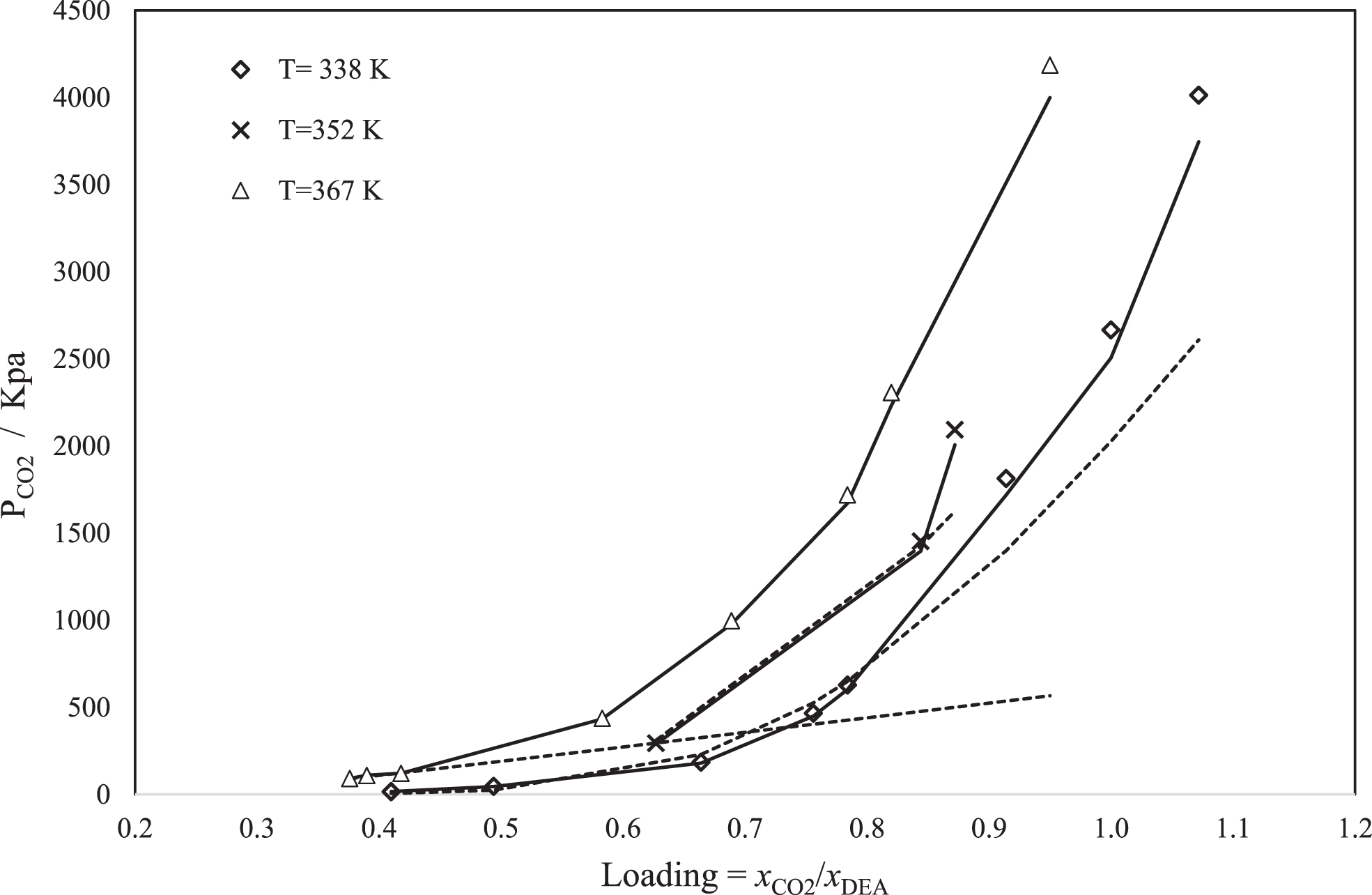
Comparison of calculated and experimental data of total pressure as a function of CO2 loading using SAFT-HR (dashed line) and PC-SAFT (Straight line), at 338 K, 352 K, 367 K, and one DEA mole fraction (0.054); lines and points represent the model results and experimental data respectively [52].
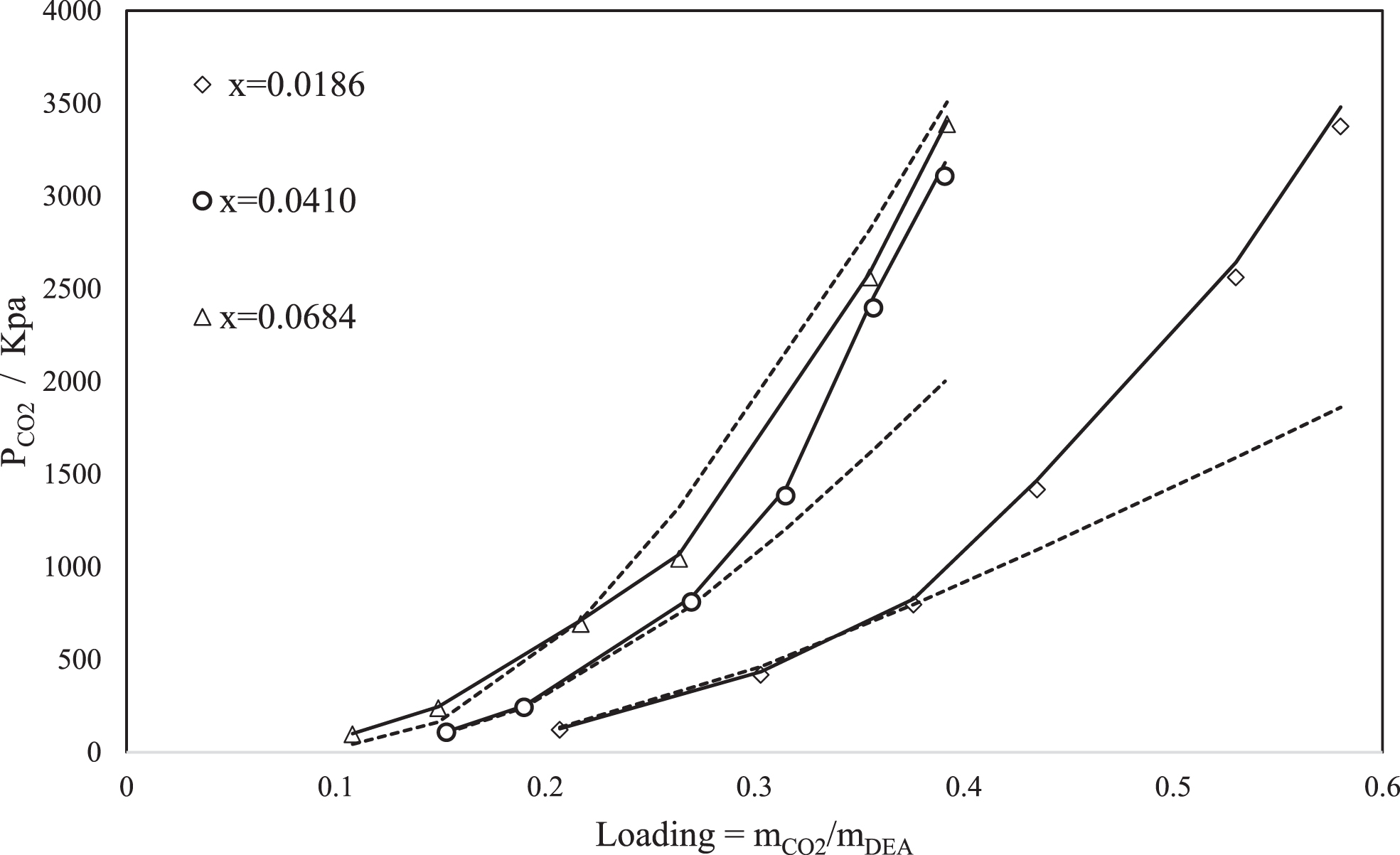
Comparison of calculated and experimental data of total pressure as a function of CO2 loading using SAFT-HR (dashed line) and PC-SAFT (Straight line), at 383 K, and three DEA mole fraction (0.0186, 0.0684 and 0.041); lines and points represent the model results and experimental data respectively [50].

Comparison of calculated and experimental data of total pressure as a function of CO2 loading using SAFT-HR (dashed line) and PC-SAFT (Straight line), at 403 K, and three DEA mole fraction (0.018, 0.068 and 0.041); lines and points represent the model results and experimental data respectively [50].

Comparison of calculated and experimental data. Total pressure as a function of CO2 loading for the CO2/MDEA+DEA system, at 300 K, 313 K, and 323 K; lines and points represent the model results and experimental data respectively [46].

Comparison of calculated and experimental data total pressure as a function of CO2 loading for the CO2/MDEA+DEA system, at 313 K, 343 K, and 393 K; lines and points represent the model results and experimental data respectively [53].
Both models are capable of reproducing the experimental solubility data of CO2 in aqueous alkanolamine solution. However, PC-SAFT model presents more accurate result since the calculation algorithms of dispersion term is highly accurate in this model.
The purpose of this article is thermodynamic modelling of the equilibrium solubility of carbon dioxide gas in aqueous solvent of diethanolamine and methyldiethanolamine in various loadings based on the PC-SAFT and SAFT-HR equations of state. Accordingly, the method of work consists of the following steps: Collecting required data (such as adjustable parameters) from articles. Determine the solubility of carbon dioxide on the MDEA aqueous solution using the method chosen in this article. Modelling the equilibrium solubility of hydrogen sulfide in MDEA aqueous solution using the SAFT-HR and PC-SAFT model.
In this work, SAFT-HR and PC-SAFT EoS is applied for the prediction of CO2-DEA-H2O ternary system in temperature range 298–478 K and CO2 partial pressure range 0.01–5,500 kPa. Because of the importance of association schemes in parameter estimation in associated EoSs, several association schemes are checked for MDEA, and the results indicate that the 3B association scheme for MDEA and DEA shows more conformity with binary VLE experimental data more than other association scheme. Therefore, both the 3B association scheme for MDEA and the 4 C association scheme for DEA (due to its consistency in CO2–H2O system) are applied. In summary, PC-SAFT EoS performs an acceptable prediction in the ternary system of CO2–MEA–H2O in a wide range of temperatures and CO2 partial pressure without using any regression in multicomponent system, and its average absolute partial pressure deviation is about 4.374% among 356 data points.