
Abstract
Quantitative structure-activity relationship study was used to investigate the relationship between anti-pfdhfr activity and structure of twenty-eight 1,3,5-triazine derivatives. We performed benchmark studies on the molecular geometry, electron properties of 1,3,5-triazine using semi-empirical(PM3), density functional theory and post Hartree-Fock methods. Followed by a QSAR study using multiple linear regression (MLR) and artificial neural networks (ANN). The QSAR models developed allow identify/describe the relationship between the biological activity of the molecules and their molecular descriptors (topological, physicochemical, electronic...). A further external set of compounds was used for validation where a high correlation between experimental and predicted anti-pfdhfr activity values is noticed. This QSAR study provides useful information for developing novel pfdhfr inhibitors. The set’s ADME properties and drug similarities, as well as newly produced compounds and reference ligand, were investigated. These findings would be extremely useful in guiding optimization for the development of new anti-pfdhfr drug candidates.
Introduction
One of the big killers of diseases in the worldwide burden is malaria [1], that is a parasitic infection spread by Plasmodium falciparum. An infected mosquito of the species Anopheles bites humans and transmits the parasite [2]. In 2018, estimates suggested by the World Health Organization (WHO) recorded 228 million cases of malaria worldwide, with 213 million cases accounting for 93% of the global burden. Nigeria is still the world’s worst-affected country, accounting for 25% of worldwide morbidity and 24% of global death [3], possibly as a result of rising resistance to present anti-Plasmodium falciparum DHFR medications, there is a need to create innovative anti-pfdhfragents. An integrated strategy for combating malaria must include prevention, vector control, and treatment using potent pfdhfrs [4]. Among the latter, which arouse growing interest among chemists are the derivatives of 1,3,5-triazines (Fig. 1) [5] are precursors for treating malaria. As a result, anti-pfdhfr activity will be a focus of our computation investigation, as will compounds derived from 1,3,5-triazine [6] that may inhibit the bifunctional pfdhfr (Plasmodium falciparum dihydrofolatereductase) enzyme’s function.

Structures of 1,3,5-triazine with atom numbering.
The in-silico prediction of anti-pfdhfr activity has been highlighted as a critical stage in the development of drugs with targeted biological activity. The computer-aided drug discovery (CADD) has proven to be a beneficial method for discovering prospective lead compounds and assisting in the development of new medications for a number of ailments [7]. In medicinal chemistry, computational analyses based on structure-based approaches, such as QSAR (Quantitative Structure Activity Relationship) method and ADME (Absorption, Distribution, Metabolism, and Excretion) proprieties are now commonly employed to assist speed up the drug design process [8–13].
QSAR has developed into a well-established area in computational drug design and drug profile exploration [14]. It’s defined as a method of identifying significant correlations between a set of structures and functions by building computational or mathematical models utilizing chemometric approaches [15]. Additionally, the drug-likeness and ADME prediction is a useful tool for selecting drug-like compounds. There are a few important considerations to remember when it comes to improving oral bioavailability in medication development. It uses to enhance data from in silico docking experiments or to prioritize hits from high-throughput screening initiatives [16].
The implementation of these three studies was the subject of this current research. The first section focuses on the development of the best QSAR models using the statically approach Multiple Linear Regression (MLR) and Artificial Neural Networks (ANN), on a set of different molecular characteristics of pfdhfr inhibitors. It would reveal fresh knowledge that might be used to design new pfdhfr inhibitors with increased potency.
A predictive QSAR model was constructed to be utilized for lead optimization and testing of novel compounds, and an in-silico evaluation of drug-likeness features was examined, which gives useful information about the activity of substances in the body that are expected to serve asinhibitors.
Computational details
Initial calculations were optimized using HyperChem 8.03 software [17]. The geometry of 1,3,5-triazine and its derivatives; were pre-optimized using the MM+force-field (rms = 0,01 Kcal/) in molecular mechanics [18]. The PM3 approach was used to completely re-optimize geometry [19].
In the next step, a parallel study has been made using Gaussian 09 program package, at various computational levels, HF/6-31++G(d,p), HF/6-311++G(d,p), B3LYP/6-31++G(d,p) and B3LYP/6-311++G(d,p) [20].
After that, the different properties of 1,3,5-triazine derivatives were calculated by using MarvinSketch 17.13.0 [21] software, ACD/Chemsketch12.0 [22] and HyperChem software(version 8.0.6) [17]. By means of these software, twenty descriptors are computed and reduced using the stepwise strategy in XLSTAT software [23] to build several QSAR models, just five descriptors of the best QSAR model have been reported in the present work.
Dataset selection
All in-vitro IC50 (μM) anti-pfDHFR activity data of twenty-eight molecules having similar structures were selected from a series of 1,3,5-triazine-based derivatives expeditiously synthesized and biologically evaluated by Gravestock et al. [5]. The negative logarithm (pIC50 = –log10 (IC50)) was used to convert all of the experimental activity IC50 values for the purpose of providing numerically greater data values, listed in Table 2, after then, it was employed as the dependent variable in the creation of the QSAR models.
QSAR modeling studies
In an attempt to determine the role of structural features of compounds, which appears to have an effect on anti-pfdhfr activity, a QSAR models were generated. The field of quantitative structure-activity relationships deals with the development of a predictive models correlating biologicalactivity (pIC50) with the physicochemical properties. Once these are available, by using statistical methods, It is possible to establish this predictive MLR-QSAR and ANN-QSAR models for a series of biologically active molecules which have shown inhibitory activity against the pfdhfr enzyme [24].
Statistical analysis and model validation
To predict the QSAR models, multiple linear regression (MLR) analysis of molecular descriptors was carried out in the present work using the stepwise strategy in XLSTATsoftware [23]. The MLR method was compared to the artificial neural network (ANN) method, which is another reliable and predictive QSAR model that is ideally suited for treating non-linear correlations between descriptors and activity [24–26]. All the ANN analyses were carried out using JMP 8.0.2.software[27].
Two basic methodologies are used to undertake external and internal validation of models [28]: internal validation utilizing the training set molecules, and external using the test set molecules by partitioning the entire data set into training and test sets; at 80% and 20%, respectively, utilizing the so-called ‘Balanced Subset Method’ (BSM) [29].
Apart from the use of fitness of several parameters, the statistical qualitative analysis of the QSAR model was validated by using the leave-one-out cross validation method (LOOCV) [30, 31]. The best model was chosen in this study using the determination coefficient (R2) and adjusted determination coefficients (
Applicability domain approach
Another pivotal issue is the definition of a locale in the compound space containing the structural, physicochemical, or natural properties data s where upon the training set of the created model is through the applicability domain (AD) and for which it is applicable to make predictions for new compounds [32, 33]. Even the most comprehensive, significant and validated models cannot reliably predict properties for all existing compounds.
Therefore, the AD of the models must be defined and only predictions for molecules falling in the training set in this domain can be considered acceptable. The method of leverage value hi for each compound i has a calculated of the QSAR model was represented (Fig. 7)
A warning should be presented when hi > h* for a test set compound, suggesting that the prediction is the result of significant model extrapolation and should not be trusted [36].
Drug likeness parameter and lipophilicity indices
A drug’s physicochemical properties of chemical compounds of series have a substantial impact on its in vivo pharmacokinetic characteristics under research with PFDHFR enzyme. The detailed analysis of drug likeness characteristics and lipophilicity indices were carried out by applying the different rules, by Lipinski’s rules [37, 38], veber’s rules [39], lipophilicity indices [40, 41], and Golden Triangle tool [42].
Results and discussion
Validation method
Equilibrium geometries of 1,3,5-triazine
The main geometrical parameters of the optimal equilibrium geometry to be employed are the most efficient theoretical strategy for the larger of 1,3,5-triazine (Fig. 1) of interest in the current study perhaps selected by comparison with experimental results.
Our investigations started by performing benchmark studies on 1,3,5-triazine using different theoretical methods (PM3, Ab-initio, DFT) in order to select the most reliable predictive method comparatively to experiment and with reduced computational cost.
Table 1 lists the main geometrical parameters of the optimized equilibrium geometry of 1,3,5-triazine are in accordance with the numbering scheme given in (Fig. 1). Table 1 lists also the corresponding experimental geometrical parameters that have been obtained by X-ray diffraction, which revealed that the molecule had D3h symmetry [43]. Since 1,3,5-triazine are planar, the calculated dihedral angle values are either 0° or 180°.
Bond lengths (in Å) and valence angles (in degree, °) of 1,3,5-triazine. Experimental data for 1,3,5-triazine are collected from Ref. [44]
Bond lengths (in Å) and valence angles (in degree, °) of 1,3,5-triazine. Experimental data for 1,3,5-triazine are collected from Ref. [44]
Optimized structures of the molecules under study [5]
(*): Test set compounds. pIC50 = -log 10 (IC50).
From the obtained values (Table 1), we can also find that the appropriate method to compute the spectroscopic parameters of the 1,3,5-triazine is the density functional theory (DFT with B3LYP/6-31++G(d,p)) which will be used to compute the quantum properties of our series of the 1,3,5-triazine derivatives.
The electrostatic potential that is created by a molecule’s electron charge density in space expands in the entire space (nuclei considered as point charges). MESP entails comprehending a variety of physical and chemical phenomena, for instance molecular reactivity, molecular recognition, intermolecular contacts, substituent effects, electrophilic reactions, and reagent-induced interactions, such as those between a drug and its cellular receptor [45]. Figure 2 shows the 3D molecular electrostatic potential surface maps (3DMESP) of 1,3,5-triazine.
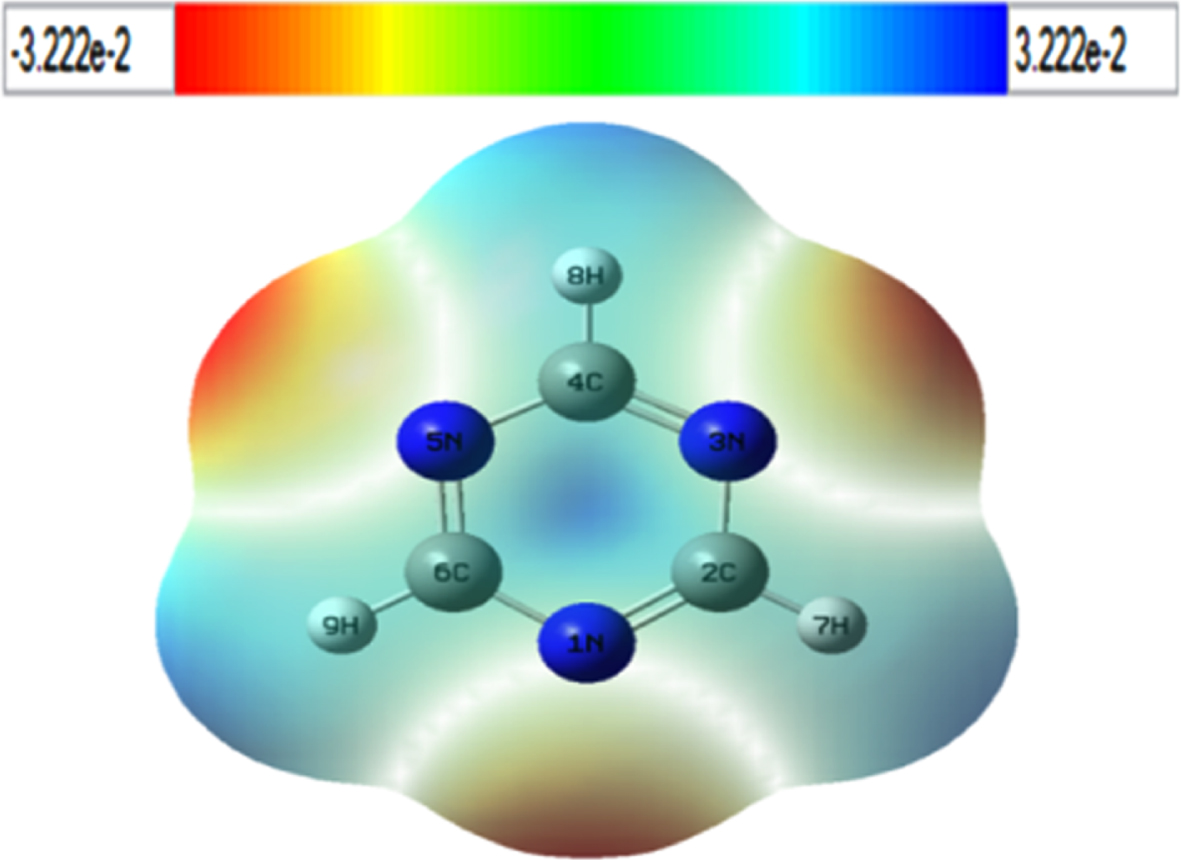
3D MESP of 1,3,5-triazine. The results are color-coded, from red (most negative) to blue (most positive).
As can be seen in Fig. 2, due to its strong electronegativity, 1,3,5-triazine exhibits negative electrostatic potentials (red zone) surrounding the nitrogen atoms (N1, N3, and N5). Additionally, we can observe positive electrostatic potentials (the blue zone) everywhere around the hydrogen and carbon atoms, which explain why these atomic sites are exposed to nucleophilic attack. Carbon atoms attached hydrogen atoms have the most positive charge per atom of hydrogen (dark blue).
In sum, 1,3,5-triazine derivatives exhibit a number of properties that may help us better understand the electrostatic interactions that may occur between the 1,3,5-triazine derivatives under study and reagents or enzyme active sites.
Multiple linear regression (MLR)
In the present study we tried to develop the statistical correlation of the best QSAR model that was derived from multi linear regression model generation (MLR).
That the physicochemical descriptors, NRB (number of rotatable bond on the molecule),
Chemical descriptors used in the regression analysis. They correspond to number of rotatable bond on the molecule (NRB (eV)), energy of highest occupied molecular orbital (E
HOMO
(eV )), energy of lowest unoccupied molecular orbital (E
LUMO
(eV )), refractive index (n) anddipolar moment (µ (D))
Chemical descriptors used in the regression analysis. They correspond to number of rotatable bond on the molecule (NRB (eV)), energy of highest occupied molecular orbital (
Among several MLR equations the best model is expressed by the following relation:
In Fig. 3, We display the experimental activity versus the predicted activity values to further created MLR model’s prediction ability.
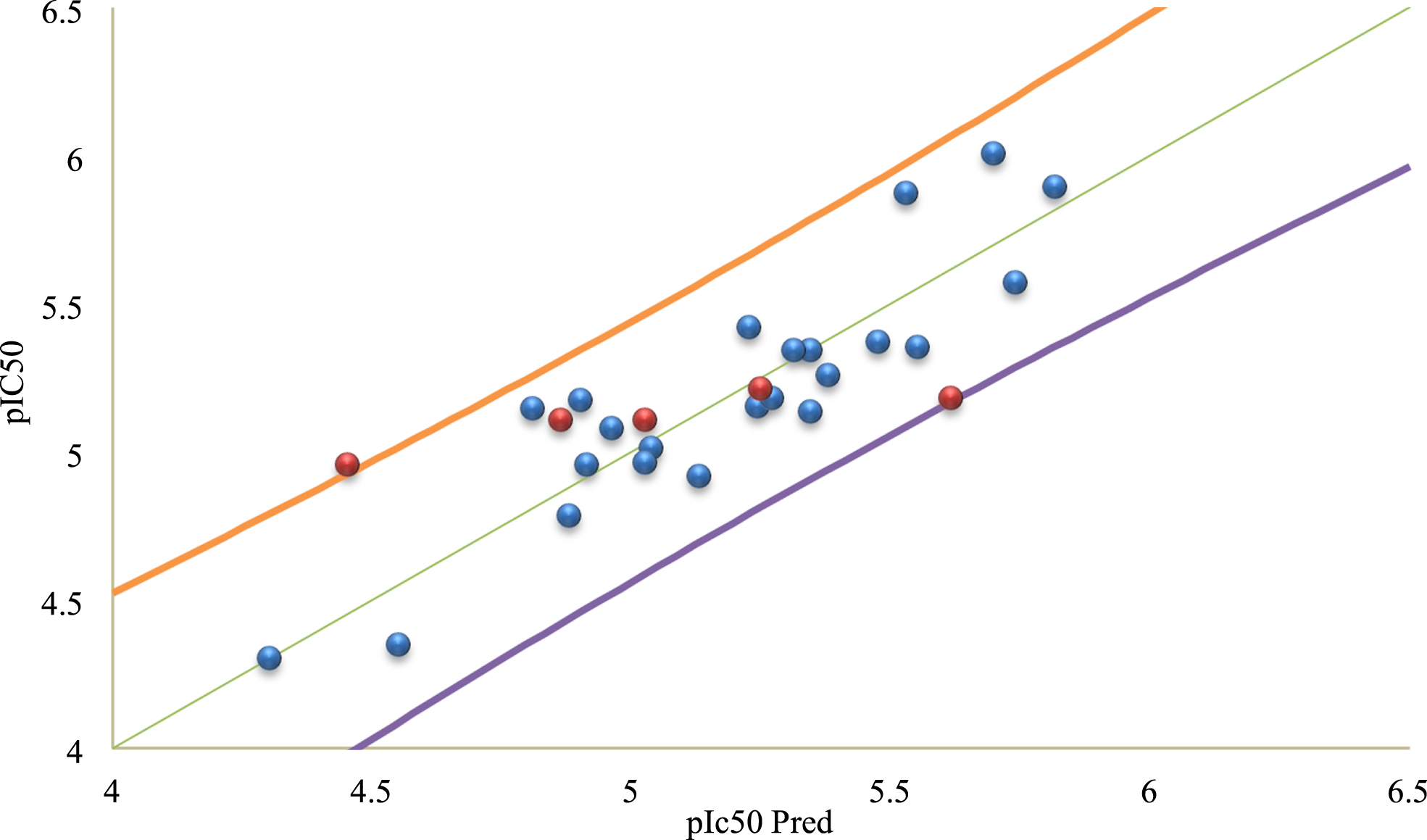
Correlations of experimental versus predicted pIC50 values using MLR.
Once developed, the model must be interpreted by analyzing all the statistical parameters. In the model obtained in Equation (1), we note that
From the observed and predicted biological activity data of the molecules given in Table 3, we can notice that there is a strong similarity between the observed and predicted pIC50 values. This can be explained by the low values of the residuals. This means that the QSAR models developed via the MLR and ANN techniques have a strong predictive capacity of the biological activity of the studied molecules according to the selected molecular descriptors (NBR
In order to detect the absence of the multi-collinearity for the selected descriptors the variance inflation factors (VIF) were calculated [46]. All five descriptors in the MLR-QSAR model have VIF values less than 5 (VIF = 1.716, 1.800, 3.023, 2.572, and 1.855, for
Moreover, the predictive power of the equation of the model is confirmed by metrics of Golbraikh and Tropsha’s criteria are listed in Table 4. The MLR model having R2 > 0.6 for both training and test sets will only be considered for validation [47]. Equation (1) exhibited high values of R2 and
MLR statistics of predicted model
This is on the other hand confirmed by metrics for external validation that has also used Golbraikh and Tropsha’s criteria to judge the predicted model from a calculation the larger
Furthermore, the robustness of the MLR model was ensuring by applying the Y-randomization test. Mostly, the Y-randomization test is used to test the stability of the predictive power of statistical models. Therefore, in the present study, this test was used to check the stability of the statistically modeled structure-activity relationship. This is to eliminate the probability of generating a QSAR model at random. We performed many Y vector random shuffles. After 100 random tests we have obtained small average values of 0.217 for
3.2.1.1. QSAR model’s applicability domain. A William’s plot is used to show the AD of models (Fig. 4). According to the “three-sigma rule” [51], the AD is established using Excel 2013 software [52], in this plot inside a square region within the standard deviation x (in this study, x = 3). Outliers are molecules with standardized residuals three times higher than the modes standard deviation.

MLR model’s applicability domain plot. The vertical dashed line represents the warning leverage (h* = 0.782), whereas the horizontal lines denote±3.
All substances in the dataset fall within the applicability range of the suggested MLR model, as can be seen by carefully examining Fig. 4. The leverage values of the inhibitors are all less than the warning h* value (0.782), and none of them exhibit standardized residuals that are more than the threshold. As a consequence, the model exhibits the best statistical parameters and strong predictive capabilities, and it can be used in this AD with a high level of confidence.
The existence of a non-linear relationship between pIC50 and the five selected descriptors by the MLR model as inputs was studied in the second stage. The number of hidden layers was determined using the value 2n+1, where n denotes the number of input layers, which plays an important role in establishing the optimum ANN architecture [53].
For pIC50 data, the architecture of the chosen ANN model was 5-3-1, with 5 descriptors in the first layer, three neurons in the hidden layer, and one neuron in the output layer after optimization. In this work, the intermediate (hidden) layer is made up of three neurons that form a deep internal pattern that identifies the strongest correlations between expected and experimental data. The output layer is made up of one neuron that returns the value of pIC50 (Fig. 5).
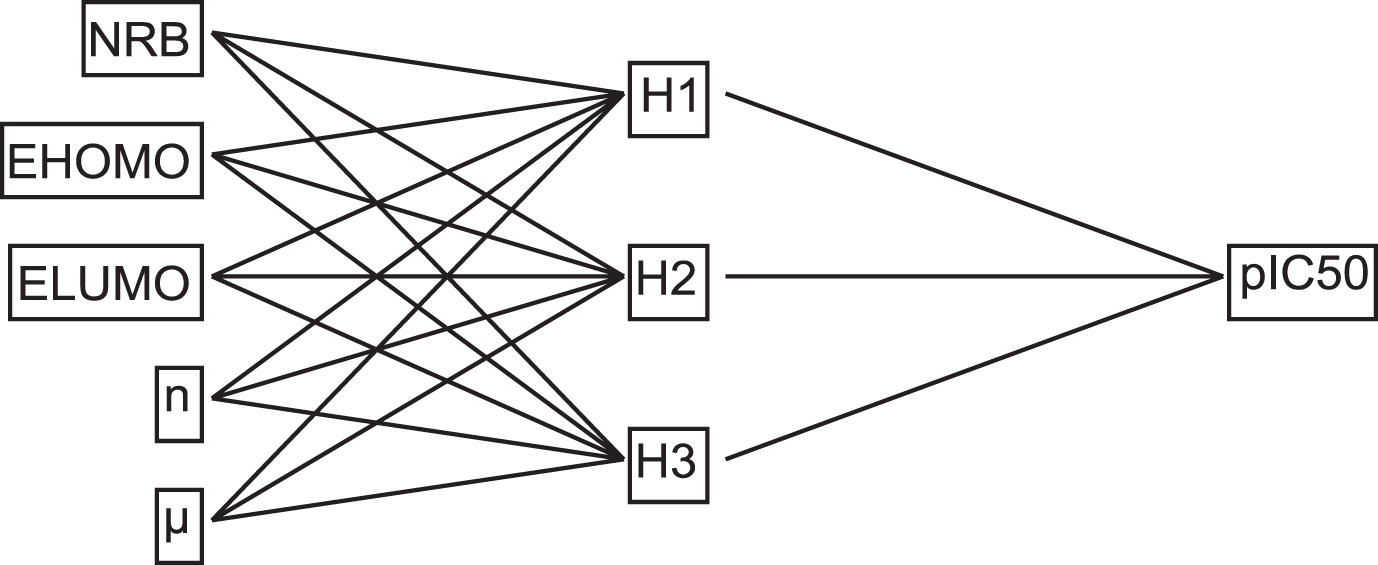
Architecture of ANN.
The Gauss Newton method was then used to train the ANN [54]. The experimental and predicted pIC50 using ANN are found to be highly correlated. This is indicated in Fig. 6 with 0.987 and 0.841 value of R2 and

Correlations of experimental versus predicted pIC50 values using ANN.

Compound 10.
We could infer that the ANN model with (5-3-1) architecture is capable of establishing a suitable link between the five descriptors and anti-pfdhfr activity based on both training and test set outcomes (Fig. 5).
A simple comparison of the values of the statistics parameters of ANN model in Table 5 with those obtained using the MLR method confirms that ANN outperforms MLR, demonstrating the existence of a non-linear relationship between the pIC50 and the five selected descriptors of the investigated compounds.
ANN and MLR statistics of predicted models
Thus, therefore, our QSAR models can be successfully applied to predict the anti-PFDHFR activity of this class of molecules.
Using the foregoing results as a guide, we made appropriate substitutions and then proceeded to calculate their activities using the proposed model Equation (1). As a result, the proposed model will help us to speed up the time when it comes to synthesizing and assessing the anti-pfdhfr activity of 1,3,5-triazine derivatives.
According to the preceding discussions, our MLR model might be used to calculate pIC50pred of various 1,3,5-triazine derivatives as shown in Table 6 and could contribute to the development of new anti-pfdhfr druglike. If we create a new compound with higher values than existing compounds, we may be able to create more active compounds than those now in use. In this manner, we performed structural alteration using compounds with the greatest pIC50 values as a template comp.10 (Fig. 7).
Values of descriptors and pIC50 for the new designed compounds (derivatives of comp.10) of the Fig. 7
Values of descriptors and pIC50 for the new designed compounds (derivatives of comp.10) of the Fig. 7
Table 6 lists the structures of the designed compounds, as well as their parameter values computed using the same procedures and the pIC50 values predicted by the MLR model.
Chemicals’ drug-likeness is a qualitative feature [55], beneficial for early-stage drug development. From the standpoint of this concept, it would be ideal to encode the equilibrium between a compound’s molecular characteristics that effects its pharmacokinetics and eventually optimizes their absorption, distribution, metabolism and excretion (ADME) in the human body like a medicine.
At present, we should evaluate the oral bioavailability of the twenty-eight 1,3,5-triazine derivatives under study. Continuous, the quickest strategy for appreciating the drug-likeness of a set is to apply “rules”, they have been applied. As first, the most commonly Lipinski’s rules are used [37, 38].
Continuous, our parameters determined that good absorption or permeation is more likely to occur when: the molecular weight (MW < 500da), number of hydrogen bond donors (HBDs < 5) (counting the sum of all NH and OH groups), to estimate hydrophobicity of molecules used the partition coefficient octanol/water (Log p < 5), and the number of hydrogen bond acceptors (HBA < 10) are all within a certain range (counting all N and O atoms). In this rule of five-score, there are a total of four violations of Lipinski’s rules.
Veber et al. identified the other two descriptors. [39]: number of rotatable bonds (NBR < 10) and topological polar surface area (PSA < 140 Å2). The TPSA is an important measure for predicting molecular transport properties, especially in the areas of blood-brain barrier (BBB) penetration and intestinal absorption [56, 57]. It is well known that molecules with a TPSA of 140 Å2 have a great ability to penetrate in an environment that is hydrophobic, like biological membranes. However, this could explain their quick penetration in hydrophilic settings, for instance, the core of transporter proteins [58]. The TPSA values were discovered to be in the range of (74,8 Å2–127,17Å2); these chemicals may be able to penetrate the BBB, resulting in increased bioavailability. TPSA was used to compute the percentage of absorption ((% ABS) in accordance with the equation the equation % ABS = 109±0.345×TPSA [59]. All of the compounds had a high % ABS, ranging from 65.126 to 83.194 %, implying that the permeability of their cellular plasmatic membrane is good.
The results obtained are shown in Table 7, they were calculated using HyperChem 8.0.8 (for MW, Log p and NH) and MarvinSketch 6.2.1 software (for HBD, HBA, NBRand TPSA). As can be seen there, The Lipinski and Veber criteria are satisfied by all substances, indicating that their theoretical oral bioavailability is optimal. The association between appropriate aqueous solubility and intestinal permeability, as well as these physicochemical molecular properties that represent the first steps in oral bioavailability.
Drug-likeness parameters and Lipophilicityindices of 1,3,5-triazine derivatives
Drug-likeness parameters and Lipophilicityindices of 1,3,5-triazine derivatives
Therefore, for the series of interest, Table 7 shows that the rule of five is 4 and 2 for Veber’s score. Indeed, to be marginal for further developments the compounds with Rule of five-scores>1 are taken into consideration [60]. Overall, our findings show that most compounds defy Lipinski and Veber rules, indicating that all chemical compounds would have no issues with oral bioavailability.
We have also defined ligand efficiency (LE), lipophilic ligand efficiency (LLE) and the golden triangle as LE = 1.4pIC50/NH, and LLE = pIC50–LogP; where NH is the number of heavy atoms [61]. They are described as crucial parameters for drug discovery, furthermore as a means of determining a compound’s potency in relation to its molecular weight. LE is influenced by ligand size, with smaller ligands having higher biological efficiency than bigger ligands on average [62, 63].
Further, we used LLE to facilitate a deeper comprehension of the affinity of structural alterations in the series, with respect to lipophilia. As a rough guide, LLE values have been stranded in the range 5–7 in drug-like space for medicinal compounds [64]. That compounds with high LE and LLE tries to improve a potency interact with biological targets [65]. In the studied series, the change of LE and LLE during optimization (Table 7).
Other characteristics that affect ADME and drug-likeness attributes such as molecular weight (MW) and distribution coefficients (logD) were used to illustrate the simultaneous absorption and clearance of optimal medicines using Warring rules and the Golden Triangle tool [42]. The Golden Triangle is a visualization tool developed from in vitro permeability, in vitro clearance and computational data designed to aid medicinal chemists in achieving metabolically stable, permeable and potent drug candidates [42]. Plotting MWvs. logDon estimated octanol: buffer (pH 7.4) and classifying compounds of a series as permeable and stable (pH 7.4).
A triangular shape known as the “golden triangle” is formed when the design properties are moved into an area with a baseline of log D7.4 = –2.0 to log D7.4 = 5.0 at MW = 200 Da and a peak at log D7.4 = 1.0 to 2.0 and MW = 450 Da, this increases the chance of success in maximizing potency, stability, and permeability. According to the fighting rule, with MW = 414 Da and logD7.4 > 1.3 have a 74% chance of 74% chance of becoming highly permeable. Golden triangle’s rules apply to the majority of our substances. These findings should aid in the development of permeability-enhancing chemicals. The metabolic stability and good membrane permeability of compounds found inside the Golden Triangle are more likely to be present than others outside.
According to the Golden Triangle (Fig. 8), the most of compounds under study are located within of it, indicating that these 1,3,5-triazine derivatives have good permeability and clearance [66]. The other compounds are the reverse, consisting of six compounds: 1,3,5,9, 20 and 22.

Permeability and clearance patterns in vitro for MW and logD.
Table 8 lists the physicochemical parameters of novel designed compounds. All of the proposed compounds’ LogP and HBA values which indicate that they have a fair absorbency and resulting in an increase in the electrostatic interactions of the 1,3,5-triazine derivatives with the amino acid residues in the active sites. They were in great accord with the most important rules of drug similarity e.g. Lipinski, Veber and Lipophilicity indices.
Drug-likeness of the new designed compounds and reference compounds
Drug-likeness of the new designed compounds and reference compounds
The number of rotatable bonds 7(NBR < 10), hydrogen bond acceptor 6 or 7 (HBA < 10) and octanol/water partition coefficients (Log p < 5) are used to forecast a compound’s lead-likeness. When compared to the reference compound 10, these compounds showed no discomfort, indicating that they have good drug likeness properties.
Starting with a comparison between the spectroscopic parameters of 1,3,5-triazine with different calculation methods showed that the B3LYP/6-31 G++(d,p) approach is sufficiently reliable. Then by studying the benchmarks on the structure and electron density of 1,3,5-triazine, we validated the use of DFT B3LYP/6-31 G++(d,p) to study the drug Likeness Screening and the structure activity-relationship for a series of 1,3,5-triazine derivatives that are known to be inhibitors of pfdhfr. The both methods MLR and ANN were used to create QSAR models. The constructed models showed a strong connection between experimental and anticipated pIC50 values, demonstrating its great predictive capacity. We have also created and proposed some novel compounds that could have a wide range of applications. As a result, this model can be used to predict the inhibitory activity of this class of compounds (1,3,5-triazine derivatives) against PFDHFR. Our work shows the use of multi-parameter optimization desirability score that the series and new compounds obey the Lipinski’s and Viber’s rules, lipophilicity indices and golden triangle rules showed that the majority of these compounds exhibit improved permeation, intestinal permeability, and oral bioavailability, as well as desirable in vitro ADME and safety properties [65].