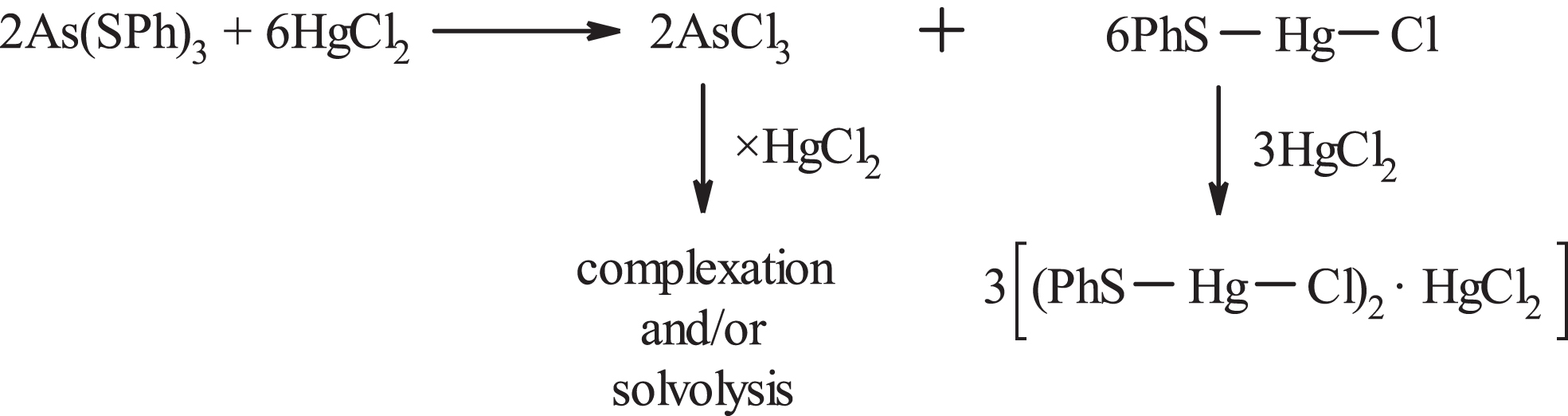The coordination ability of As(III) thiolates, having two kinds of Lewis base atoms [As(III) and S(II)], towards heavy metal salts was virtually not studied. We have found that As(SPh)3 (A), Me-As(SPh)2 (B), and 2-O2N-PhS-As(SPh)2 (C) with the acetates M(OAc)2 (M = Cd, Pb, Hg) gave the metal thiophenolate M(SPh)2 (except in the case of B and Cd(OAc)2 which gave PhS-Cd-OAc·Cd(SPh)2) and with HgCl2 gave (PhS-Hg-Cl)2·HgCl2. The thiophenolates of cadmium and lead(II) were not reactive towards their corresponding acetates and chlorides, while Hg(SPh)2 gave PhS-Hg-OAc and (PhS-Hg-Cl)2·HgCl2. Likely mechanisms for these reactions are suggested.
Monodentate, non-fissionable As(III) compounds like aliphatic and aromatic arsines (R3As) do form complexes with heavy metal cations like Cd(II) [9] and Hg(II) [6, 9] where a bond As:⟶M is formed using the stereochemically and chemically active lone electron pair of As(III).
The composition of the complexes depends mainly on the initial stoichiometries of arsines and cations used and not on the nature of ligands bound to As(III) or to the anions bound to the metal cation [6, 9].
It is noteworthy that HgX2 forms a greater variety of complexes compared to CdX2 with arsines due to ability of HgX2 to form oligomeric chains (HgX2)y [9].
In a review on the thiolates of As(III) [12] it was noted that only two publications mentioned the interaction of these thiolates with HgCl2. The first [4] mentioned that HgCl2 forms a white solid with As(SPh)3 in benzene. In the second publication [21] it was stated that various diphenyl aryldithioarsonites and one phenyl diarylthioarsinite with HgCl2 in ethanol gave insoluble solids of the type Ar-As(SPh)2·4HgCl2, a quite unexpected result.
Trithioarsenites, dithioarsonites and thioarsinites of arsenic have two different nucleophilic atoms, As(III) and S(II), which can be, under proper conditions, chemically active towards metal cations.
In the thiolates of As(III), the As-S bond is reactive and can be broken by e.g. H2O, I2, O2, H2O2 [12]. They react, although in a complicated way, with the electrophilic RCOCl and (RCO)2O; As(SPh)3 does not react with RX, but Et-As(SR)2 reacted with MeI giving Et-AsI2 and Me2S+-R I-, where the nucleophile is the S(II) and not the As(III) atom [12].
PhS-As(SPh)2 (A) Me-As(SPh)2 (B) (C)
Cd(OAc)2·2H2O (I) Pb(OAc)2·3H2O (II) Hg(OAc)2 (III) HgCl2 (IV)
In this paper we report on the reaction of the triphenyl trithioarsenite (A), diphenyl methyldithioarsonite (B), and diphenyl 2-nitrophenyldithioarsonite (C) with Cd(OAc)2·2H2O (I), Pb(OAc)2·3H2O (II), Hg(OAc)2 (III), HgCl2 (IV), and on A plus BiCl3. Under various stoichiometries and conditions the products obtained were formulated as 1–6. We used the acetates hoping to facilitate the identification and establish the composition of the products with IR and 1H-NMR, and we found that in all cases the attacking atom was S(II) and not As(III). The latter atom under certain condition can also react with III giving minute amounts of elemental mercury
Experimental section
Materials and methods
Triphenyl trithioarsenite, As(SPh)3 (A), was prepared from As2O3 and non-distilled thiophenol [20] and by 1H-NMR contained 10% PhSSPh that was present in thiophenol. Diphenyl methyldithoarsonite, Me-As(SPh)2 (B), was prepared from methylarsonic acid and non-distilled thiophenol. Because it was not chromatographed [23] it contained 20% PhSSPh. Diphenyl 2-nitrophenyldithioarsonite, 2-O2N-Ph-As(SPh)2 (C), was prepared as described [23], the PhSSPh being extracted by petroleum ether. M.p. 92–93 °C (lit. 92–93 °C [23]). Mercury(II) thiophenolate, Hg(SPh)2 (3) was obtained from mercuric acetate and thiophenol [10], m.p. 153–154 °C (lit. 150 °C [1]). The other metal salts were Analytical Reagent grade.
Most reactions were run in centrifuge tubes (scales 50μmol to 300μmol) and in some cases in 10 mL round-bottomed flasks. In all cases the solids were washed in centrifuge tubes by stirring for at least 30 min each time.
The progress of the reactions was followed by thin chromatography (TLC), using microslides coated with silica gel 60 H (Merck) and visualization was effected by iodine vapors. The unreactive PhSSPh, was a useful internal marker for comparing the consumption of A, B and C. Melting points were determined on an Electrothermal, model 9100, melting point apparatus. Infrared spectra were taken in KBr discs on a Thermo Fisher Scientific, model Nicolet iS20, FT-IR spectrometer. 1H-NMR spectra were obtained using a Bruker Ascent-TM, Avance III HD 600 MHz instrument, using TMS as an internal standard. Electrospray ionization mass spectra (positive mode) were obtained using an Applied Biosystems instrument. Elemental analyses were performed in the FSE Microanalysis Laboratory, University of Manchester, England.
The reaction of As(SPh)3 (A) with salts of heavy metal cations I-IV
The reaction of the methanol-insoluble triphenyl trithioarsenite (A) with the methanol-soluble Cd(OAc)2·2H2O (I) gave the cadmium(II) thiophenolate (1), Table 1, insoluble in MeOH and soluble in warm DMSO from which did not precipitate at room temperature (RT). IR (KBr): 3053 w, 1577 m, 1475 s, 1437 s, 1082 s, 1022 s, 735 vs, 686 vs, 476 m.
Reaction conditions, products, yield and melting points for the reaction of As(III) thiolates with acetates of Cd(II), Pb(II) and Hg(II) and HgCl2. Metal salt: Cd(OAc)2·2H2O (I), Pb(OAc)2·3H2O (II), Hg(OAc)2 (III), HgCl2 (IV)
As(III)
Metal salt
Molar ratio As(III)/M
Solvent
Reaction time, h
Precipitated product
Washed with
Yield
Melting point, °C
As(SPh)3 (A)
I
1 : 1
MeOH
4
Cd(SPh)2 (1)
MeOH
85
>300a
II
1 : 1
MeOH
4
Pb(SPh)2 (2)
MeOH
94
190–192b
III
1 : 1
MeOH
4
Hg(SPh)2 (3)
MeOH
95
151–153c
IV
1 : 1 ⟶ 1 : 5
CHCl3/Me2CO 1 : 1 v/v
1–25
(PhS-Hg-Cl)2·HgCl2 (6)
Me2CO
9–100
195–197
Me-As(SPh)2 (B)
I
1 : 4
Et2O/MeOH 1 : 2 v/v
30
PhS-Cd-OAc·Cd(PhS)2 (4)
MeOH, Et2O
50
304–306
II
1 : 4
Et2O/MeOH 1 : 2 v/v
72
Pb(SPh)2 (2)
MeOH, Et2O
28
191–193b
III
1 : 1
Et2O/MeOH 1 : 2 v/v
3
Hg(SPh)2 (3)
MeOH, Et2O
83
151–153c
IV
1 : 3
Et2O
5
(PhS-Hg-Cl)2·HgCl2 (6)
Et2O
86
197–199
(C)
I
1 : 2
Et2O/MeOH 1 : 2 v/v
24
Cd(SPh)2 (1)
MeOH, Et2O
82
>300a
II
1 : 3
Et2O/MeOH 1 : 2 v/v
24
Pb(SPh)2 (2)
MeOH, Et2O
39
190–192 b
III
1 : 1
Et2O/MeOH 1 : 2 v/v
2
Hg(SPh)2 (3)
MeOH, Et2O
64
153–155c
IV
1 : 5
Et2O
3
(PhS-Hg-Cl)2·HgCl2 (6)
Et2O
70
197–199
aLit.: 339–344 °C [5]; bLit.: 195–196 °C [21]; cLit.: 150 °C [1], 153–4 °C [10].
The reaction of a solution of Pb(OAc)2·3H2O (II) in methanol with A gave the lead(II) thiophenolate (2), Table 1, as a very pale yellow solid, soluble in DMSO at RT. IR (KBr): 3425 w, broad, 3050 w, 1574 m, 1473 s, 1435 s, 1083 m, 1068 w, 1022 m, 733 vs, 688 vs, 474 m.
Adding solid A to a solution of Hg(OAc)2 (III) in MeOH, the mercury(II) thiophenolate (3) was produced under 1 : 1 molar ratio, Table 1, soluble in warm DMSO. IR (KBr): 3047 w, 1576 w, 1470 w, 1435 s, 1302 w, 1084 m, 1066 m, 1022 s, 1001 m, 904 m, 784 s, 737 vs, 688 vs, 486 s. 1H-NMR (DMSO-d6, TMS), δ: 7.08 (t, J 7.8 Hz, 2 H, para-H), 7.16 (t, J 7.8 Hz, 4 H, meta-H), 7.37 (d, J 7.2 Hz, 4 H, ortho-H).
To solutions of A in CHCl3/Me2CO 1 : 1 v/v were added dropwise solutions of HgCl2 (IV) in CHCl3/Me2CO 1 : 1 v/v at molar ratios 1 : 1, 1 : 2, 1 : 3 1 : 4 and 1 : 5 and the suspensions stirred at RT for 4, 4, 3, 1 and 1 h, respectively. The solids isolated [9–100% as (PhS-Hg-Cl)2·HgCl2 (6)] were free of As2O3 and they had the same solubilities: insoluble in CHCl3, MeOH, CHCl3/Me2CO 1 : 1 v/v, insoluble in Me2CO at RT but soluble in boiling Me2CO from which precipitated on cooling to RT, and soluble in DMSO (see also Section 2.6). All samples decomposed at 195–197 °C, Table 1. IR (KBr): 3047 w, 1572 w, 1475 w, 1433 m, 1068 w, 1020 w, 910 w, 739 vs, 685 vs, 484 s.
The reaction of Me-As(SPh)2 (B) with salts of heavy metal cations I-IV
The reaction of diphenyl methyldithioarsonite (B) (0.225 mmol) with Cd(OAc)2·2H2O (II), Table 1, was incomplete by TLC and gave a white solid [46 mg, 50% as PhS-Cd-OAc·Cd(SPh)2 (4)] insoluble in hot MeOH and in hot DMSO, that on heating turned light brown at 299 °C and frothed at 304–306 °C. Calculated for PhS-Cd-OAc·Cd(SPh)2, C20H18O2S3Cd2 (Mr 611.33): C 39.29, H 2.97, S 15.73, Cd 36.77% ; found: C 37.19, H 2.78, S 15.78, Cd 36.06% . IR (KBr): 3450 w, broad, 3053 w, 1574 s, 1552 s, 1475 s, 1433 m, 1402 m, 1336 w, 1080 m, 1022 m, 860 m, 820 s, 737 vs, 690 vs, 623 w, 474 m.
With Pb(OAc)2·3H2O (II), B gave low yields of Pb(SPh)2 (2) even after 3 days reaction, and mercuric thiophenolate (3) was produced in 83% yield from Hg(OAc)2 (III) and B under 1 : 1 molar ratio, Table 1.
The reaction of B and HgCl2 (IV) under 1 : 3 stoichiometry in Et2O gave 86% (PhS-Hg-Cl)2·HgCl2 (6) as the insoluble product, Table 1. Its m.p. and IR(KBr) was the same as the product from the reaction of A and HgCl2. 1H-NMR (DMSO-d6, TMS), δ: 7.09 (apparent t, J 7.2, 7.8 Hz, 2 H, para-H), 7.18 (apparent t, J 7.2, 7.8 Hz, 4 H, meta-H), 7.39 (d, J 7.2 Hz, 4 H, ortho-H). Calculated for (PhS-Hg-Cl)2·HgCl2, C12H10Cl4S2Hg3 (Mr 961.92): C 14.98, H 1.05, S 6.67% ; found: C 15.14, H 1.03, S 6.68% .
The reaction of 2-NO2-Ph-As(SPh)2 (C) with salts of heavy metal cations I-IV
The reaction of C with Cd(AcO)2·2H2O (I) gave Cd(SPh)2 (1) in 82% yield, but with Pb(OAc)2·3H2O (II) low yields of Pb(SPh)2 (2) were obtained, while under 1 : 1 molar ratio of C and Hg(OAc)2 (III) the mercuric thiophenolate (3) was isolated in 64% yield, Table 1.
C reacted with an excess of HgCl2 in Et2O giving 70% (PhS-Hg-Cl)2·HgCl2 (6). Its solubilities and IR (KBr) spectra were the same as in the case of A or B + HgCl2 and its 1H-NMR (DMSO-d6, TMS) showed only the PhS-protons. From warm acetonitrile, 6 gave microcrystals not suitable for X-ray studies, while from warm acetone the crystals were better but still not suitable for X-ray analysis.
The reaction of As(SPh)3 (A) with BiCl3
The reaction of A with BiCl3 (1 : 1 molar ratio) in an oven-dried centrifuge tube in dried CH2Cl2/Me2CO 1 : 3 v/v slowly (3 days) deposited a white solid that was As2O3 [by IR (KBr), 804 cm–1] indicating 42% decomposition of As(SPh)3.
The redistribution (scrambling) reaction of thiophenolates of Cd(II), Pb(II) and Hg(II) with their corresponding acetates and chlorides
The redistribution reaction conditions and the solids isolated are shown in Table 2, where the Cd(SPh)2 (1) and Pb(SPh)2 (2) were not reactive.
Redistribution (scrambling) reactions of M(SPh)2 with M(OAc)2·xH2O and MCl2·yH2O (M = Cd, Pb, Hg)
Thiolate
Acetate or chloride
Molar ratio
Solvent
Reaction time
Isolated solid
Yield or Recovery, %
Melting Point °C
Cd(SPh)2 (1)
Cd(OAc)2· 2H2O (I)
1 : 5
MeOH
15 h
Cd(SPh)2 (1)
73
>300a
CdCl2·H2O
1 : 1
H2O
4 days
Cd(SPh)2 (1)
103
>300a
Pb(SPh)2 (2)
Pb(OAc)2· 3H2O (II)
1 : 5
MeOH
15 h
Pb(SPh)2 (2)
103
193–194b
PbCl2
1 : 1
H2O
4 days
Pb(SPh)2 (2)
101
190–194b
Hg(SPh)2 (3)
Hg(OAc)2 (III)
1 : 1
CHCl3/MeOH 1 : 1 v/v
1 h
PhS-Hg-OAc (5)
92
134c
HgCl2 (IV)
1 : 1⟶ 1 : 4
CHCl3/Me2CO 2 : 1 v/v
4 h
(PhS-Hg-Cl)2· HgCl2 (6)
46–58
196–197
aLit.: 339–344 °C [5]; bLit.: 195–196 °C [21]; cEtS-Hg-OAc has the same m.p. (134 °C) [2].
In the case of Hg(SPh)2 (3) and Hg(OAc)2 (III) (1 : 1 molar ratio) the solution obtained in CHCl3/MeOH 1 : 1 v/v was evaporated and dried in vacuo to give a glass. After trituration with ether, a white slightly hygroscopic solid was obtained, formulated as PhS-Hg-OAc (5) based on its 1H-NMR spectrum, that was sparingly soluble in Et2O, moderately soluble in CHCl3, CH2Cl2 and MeOH. In DMF and DMSO gave clear colorless solutions, while pure Hg(OAc)2 in DMF gave a yellow solution and in DMSO gave an orange solid and a yellowish supernatant. At 134 °C melted to mobile opalescent oil. IR (KBr): 3385 mw, broad, 3043 w, 1576 s, 1550 s, 1510 s, 1406 s, 1333 m, 1076 w, 1018 s, 930 m, 742 vs, 668 vs, 663 s, 617 m, 478 m. 1H-NMR (DMSO-d6, TMS), δ: 1.93 (s, 3 H, CH3COO), 7.07 (apparent t, J 7.2, 7.8 Hz, 1 H, para-H), 7.16 (apparent t, J 7.8 Hz, 2 H, meta-H), 7.42 (d, J 7.2 Hz, 2 H, ortho-H). ESI (positive mode) MS: 110 (60%), 125.26 (100%), 186.22 (28%), 218.22 (55%), 327.18 (8%), 389.09 (9%), 528.92 (12%), 728 (4%).
The reaction of 3 with HgCl2 (IV), under 1 : 1, 1 : 2, 1 : 3 and 1 : 4 molar ratios, in CHCl3/Me2CO 2 : 1 v/v for 4 h gave suspensions that were evaporated, dried and extracted with Me2CO (in order to extract any free HgCl2) to give (PhS-Hg-Cl)2·HgCl2 (6) in 57, 50, 58 and 46% yields. The solids were insoluble in MeOH, CHCl3 and MeCN, sparingly to moderately soluble in Me2CO and very soluble in DMSO (see also Section 2.2). Recrystallization from Me2CO gave poor quality crystals. The solids melted at 196–197 °C giving turbid oils and had the same IR spectra. IR (KBr): 3047 w, 1572 w, 1473 w, 1433 m, 1072 w, 1020 w, 739 vs, 688 vs, 484 m. ESI (positive mode) MS: 110 (25%), 125.13 (42%), 186.16 (48%), 218.22 (100%), 327.18 (15%), 389.09 (2%), 528.79 (19%), 728.07 (5%).
Results and discussion
The reaction of As(SPh)3 (A) with heavy metal salts I-IV
With I, II and III under 1 : 1 molar ratios
The trithioarsenite (A) did not form an adduct with the acetates M(AcO)2 (I)–(III) under 1 : 1 molar ratios in methanol but rather it reacted forming the methanol-insoluble thiophenolates (1)–(3), Table 1, according to the Equation (1).
The other product should be PhS-As(OAc)2 and not As(OAc)3 because in the latter case some As(SPh)3 should have been present but TLC (petroleum ether) showed that A had all reacted.
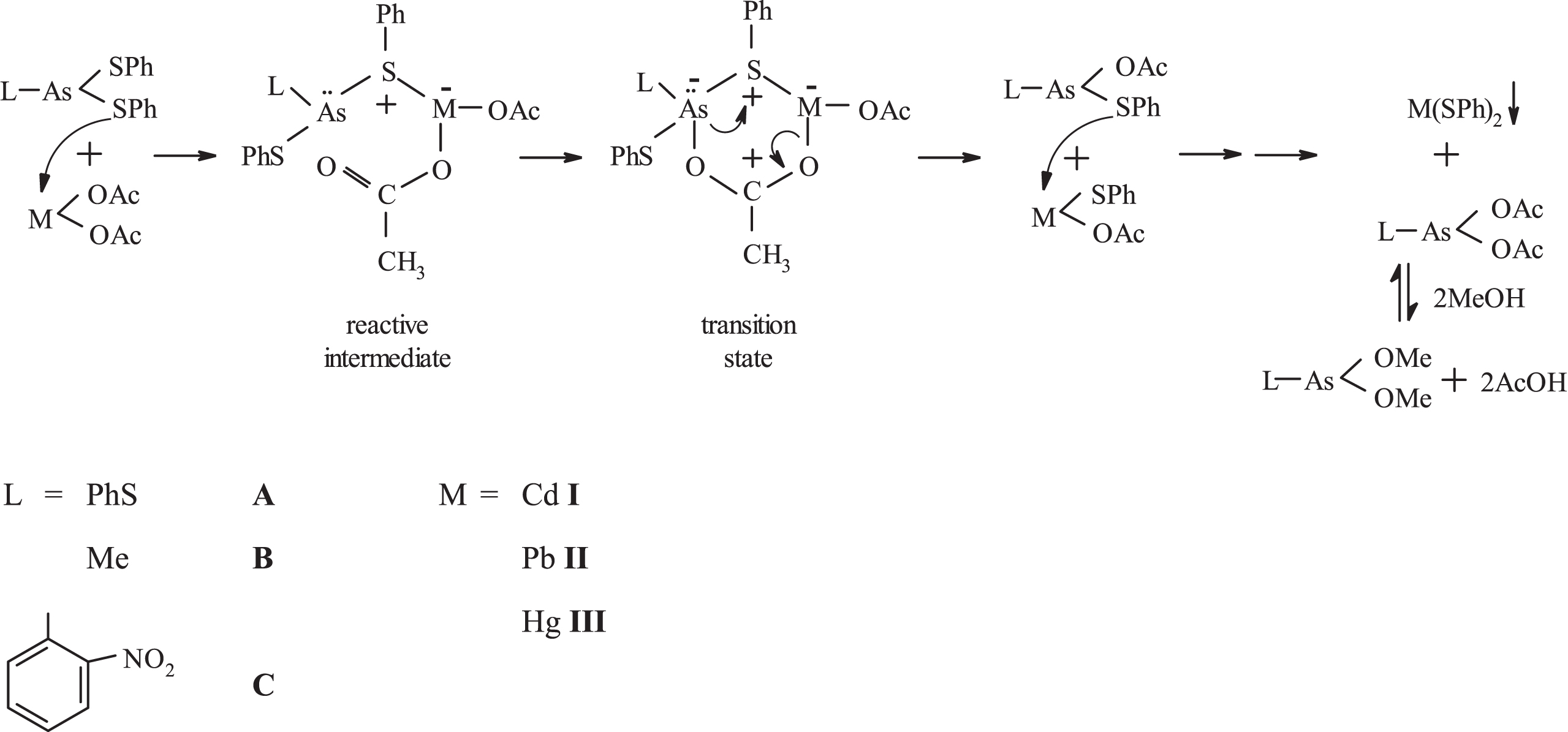
A plausible mechanism is shown in Figure 1, where the attacking atom is not the As(III) but the S(II). The Lewis adduct, being a reactive intermediate, decomposes probably via the 6-memberd transition state shown giving PhS-M-OAc that reacts further giving M(SPh)2 and L-As(OAc)2. The acetate PhS-As(OAc)2 in MeOH should be in equilibrium with PhS-As(OMe)2 and AcOH. The latter was detected (smell) after evaporating the methanolic supernatant and washing.
A plausible mechanism for the reaction of L-As(SPh)2 (L = PhS, Me and 2-nitrophenyl) with M(OAc)2 (M = Cd, Pb and Hg) under 1 : 1 molar ratio in methanol-containing solvents.
With excess I, II and III
In MeOH, under 1 : 5 molar ratio of A/Cd, 82% Cd(SPh)2 (1) was obtained and under 1 : 4 molar ratio of A/Pb, 87% Pb(SPh)2 (2) was isolated (not shown in Table 1). The thiophenolates 1 and 2 are not reactive towards I and II (Section 3.5) and, therefore, Equation (2) describes the reactions.
Only under 1 : 1 molar ratio of As(SPh)3 (A) with Hg(OAc)2 (III) in MeOH the thiophenolate Hg(SPh)2 (3) was produced in 95% yield according to Equation (1). With excess (III) both PhS-As(OAc)2 and Hg(SPh)2 of Equation (1) should be reactive. The former giving As(OAc)3 and more Hg(SPh)2, Equation (2), and the latter producing PhS-Hg-OAc (5) (see Section 3.6). In this case, Equation (3) should describe the reaction. Thus, running the reaction under 1 : 3 molar ratio of A/III in CHCl3/MeOH 1 : 3 v/v the product 5, expected based on Equation (3), was obtained in 88% yield but contaminated with As2O3 [by IR (KBr), 808 cm–1]. Attempts at the preparation of pure 5 by varying the mode of addition of the reagents and the reaction time (15 min ⟶ 24 h) the formation of As2O3 could not be avoided. The As2O3 should originate from hydrolysis of As(OAc)3 because it is known [7] that it hydrolyzes by moist air to AcOH and As(OH)3 (which spontaneously dehydrates to As2O3). However, it has been reported that Ph-As(OAc)2 requires heating in H2O and let it stand for several days in order to give (Ph-AsO)x [15].
With IV under 1 : 1 ⟶ 1 : 5 molar ratios
The product (PhS-Hg-Cl)2·HgCl2 (6) free of As2O3 was obtained only when the reactants, As(SPh)3 (A) and HgCl2 (IV) under 1 : 1, 1 : 2, 1 : 3, 1 : 4 and 1 : 5 molar ratios, have been dissolved in CHCl3/Me2CO 1 : 1 v/v as described in the experimental section. However, when IV in Me2CO was added to A in CHCl3 and stirred at RT for 30 min to 18 h, the product was contaminated with As2O3.
The reaction of A with IV under 1 : 1 to 1 : 5 stoichiometries gave (PhS-Hg-Cl)2·HgCl2 (6) in yields 9, 29 and 53% calculated on Equation (4) and 69 and 100% calculated on Equation (5).
By TLC we detected As(SPh)3 only in the 1 : 1 and 1 : 2 cases indicating that the intermediates (PhS)2As-Cl and PhS-AsCl2 were more reactive than As(SPh)3 towards HgCl2 (IV) and should afford AsCl3.
Coordination of the monomer PhS-Hg-Cl to HgCl2 to give (PhS-Hg-Cl)2·HgCl2 (6) is not unreasonable although previous preparations showed that t-BuS-Hg-Cl [3] and PhSe-Hg-Br [16] prefer to cyclize and, moreover, add Lewis bases.
On the other side, AsCl3, by analogy with arsines [9], can probably coordinate with HgCl2 to give, for example, (AsCl3)2·HgCl2 (class A), (AsCl3)2·(HgCl2)2 (class B) or (AsCl3)2·(HgCl2)4 (class D in the classification of Evans et al. [9]). There are no literature reports on complexes AsCl3·yHgCl2 or As2O3·zHgCl2 (although reaction between Ar-As(OR)2 and Ar2As-OR and HgCl2 has been reported [14]). Attempts to isolate complexes of AsCl3 and HgCl2 by working up the supernatants were unsuccessful because the solids obtained contained (by IR) As2O3.
There are two conceivable ways by which PhS-Hg-Cl can be formed. The first involves the prior formation of AsCl3 and the insoluble in CHCl3/Me2CO thiophenolate Hg(SPh)2, both of which can react with HgCl2, Figure 2. This route is unlikely because Hg(SPh)2 should be formed via PhS-Hg-Cl (cf Figure 1). The second mechanism, Figure 3, involves the stripping of one PhS- group each time from As(SPh)3, Cl-As(SPh)2 and Cl2As-SPh by a free HgCl2 to give AsCl3 and PhS-Hg-Cl which by complexation with HgCl2 gives 6. This mechanism is more reasonable and also explains the non-detection, by TLC, of the intermediates Cl-As(SPh)2 and Cl2As-SPh.
A not likely mechanism for the formation of (PhS-Hg-Cl)2·HgCl26 from As(SPh)3 and HgCl2via PhS-Hg-SPh.
A more likely mechanism for the formation of (PhS-Hg-Cl)2·HgCl26 from As(SPh)3 and HgCl2via PhS-Hg-Cl.
The reaction of Me-As(SPh)2 (B) with heavy metal salts I-IV
The reaction of excess Cd(OAc)2·2H2O (I) with B was incomplete (by TLC) after 30 h stirring. The solid that was isolated decomposed at 304–306 °C, it was insoluble even in hot DMSO, thus preventing running a 1H-NMR spectrum, but its elemental analyses indicated that it was PhS-Cd-OAc·Cd(SPh)2 (4) arising as per Equation (6). Its IR (KBr) spectrum showed PhS- and AcO- groups and additional two bands at 860 m and 820 s cm–1 that could not be assigned.
The dithioarsonite (B) also differed from the trithioarsenite As(SPh)3 (A) in its reaction with Pb(OAc)2·3H2O (II) where B gave poor yields of Pb(SPh)2, Table 1, in an incomplete (by TLC) reaction even with excess II. The reason for the differences in the behavior of BversusA towards Cd(OAc)2·2H2O (I) and Pb(OAc)2·3H2O (II) may be due to the electron-donating ability of the Me-group that can force the As(III) to interfere with the attack of S(II) at these metal cations or to reluctance at attaining the transition states, Figure1.
When to a solution of B in Et2O was added very slowly a solution of Hg(OAc)2 (III) (1 : 1 molar ratio) in MeOH then 83% Hg(SPh)2 (3) was isolated, Table 1, a result that can be attributed to the greater thiophilicity of Hg(II) compared to Cd(II) and Pb(II). When the mode of addition was reversed then, after about half of Me-As(SPh)2 had been added, the produced white 3 turned gray due to formation of elemental mercury. The mercury can be seen as a black solid after dissolution of Hg(SPh)2 in warm CHCl3. The reduction of Hg(II) to Hg0 must be accompanied by oxidation of, most likely, As(III) to As(V), implying attack of As(III) at Hg(OAc)2 and/or PhS-Hg-OAc.
The reaction of B with HgCl2 (IV) (1 : 2 molar ratio) in Et2O was incomplete (by TLC) probably indicating inactivation of HgCl2 by coordination. However, under 1 : 3 molar ratio the reaction was complete and gave 86% of the adduct (PhS-Hg-Cl)2·HgCl2 (6), Table 1, according to Equation (7). Working up the ether supernatant and washings a solid was obtained that by 1H-NMR (DMSO-d6) had no Me-group implying that the volatile (b.p. 132–133 °C [17]) Me-AsCl2 has been evaporated, indirectly indicating that under these conditions Me-AsCl2 did not coordinate with HgCl2. The IR (KBr) spectrum of the solid showed the presence of As2O3 (804 s cm–1) indicating C-As bond breakage. How this happened is not known, but C-As bond breaking under very mild conditions are known [13, 24]. However it has been reported that Ph-AsCl2 and HgCl2 (1 : 1 molar ratio) in EtOH do not split Ph-group in the course of 12 h [18].
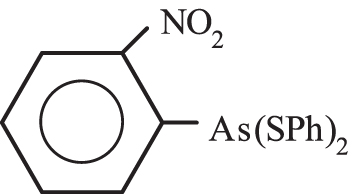
The reaction of 2-O2N-Ph-As(SPh)2 (C) with heavy metal salts I-IV
The ortho nitrophenyl group in C is electron-withdrawing and it is expected that the As(III) and to a lesser extend the two S(II) atoms to be less nucleophilic towards heavy metal cations, compared to the methyl group in B. The results showed that from C and Cd(II) or Pb(II) the metal thiophenolates 1 and 2 were obtained with good and low yields, respectively, Table 1.
The mercuric thiophenolate (3) was obtained in moderate yields when a solution of Hg(OAc)2 in MeOH was added slowly (90 min) to a solution of C in Et2O. When the mode of addition was reversed, then the Hg(SPh)2 obtained (75%) was gray, i.e. contaminated by elemental mercury probably indicating attack of As(III) at Hg(OAc)2 and/or PhS-Hg-OAc.
The variation of the yields obtained from the reaction of A, B and C with metal acetates, Table 1, is difficult to explain solely on the electron donating or withdrawal ability of the substituents of As(III) (PhS-, Me-, Ar-) which is expected to affect the formation (stability) and collapse of the Lewis adducts initially formed, Figure 1.
Early in 1933 [19] it was found that the reaction of 2-O2N-Ph-As(SPh)2 (C) (and other monosubstituted diphenyl aryldithioarsonites) with HgCl2 in ethanol gave insoluble compounds formulated as R-As(SPh)2·4HgCl2. Only one [4-O2N-Ph-As(SPh)2] melted at 196 °C, the other solids sublimed in the temperature range 190–230 °C.
We found that the reaction of C and HgCl2 (IV) (1 : 5 molar ratio) in Et2O was complete (by TLC) in≤3 h at RT and gave 70% (PhS-Hg-Cl)2·HgCl2 (6), Equation (7), while under 1 : 3 molar ratio in Et2O/MeOH 2 : 1 v/v the yield of 6 was only 46% . Thus, we believe that the ethanol-insoluble products obtained by Schuster [19] were mixtures of (PhS-Hg-Cl)2·HgCl2 and (Ar-AsO)x, the latter arising from hydrolysis of the chlorides Ar-AsCl2.
The reaction of BiCl3 with As(SPh)3 (A)
If BiCl3 had reacted with A the orange Bi(SPh)3 [11], should have been obtained. However, under dry conditions the reaction very slowly gave As2O3 arising from hydrolysis of A probably catalyzed by HCl [12] formed from BiCl3 and PhSH.
The redistribution (scrambling) of ligands between M(SPh)2 and M(OAc)2 or MCl2 (M = Cd, Pb, Hg)
Compounds of the type RS-Hg-OAc have been prepared by reacting Hg(SR)2 and Hg(OAc)2 (1 : 1 molar ratio) in water (for R = Me, Et [2]) or MeCN (for R = Ph [22]), while compounds of the type RS-Hg-Cl have been prepared by reacting HgCl2 with equimolar amounts of RSH in ethanol (R = Me, Et) [2]).
As noted before, the products obtained from the reaction of A, B and C with the metal acetates I, II and III under 1 : 1 molar ratios were the metal thiophenolates M(SPh)2 (except in the case B + I), Table 1, arising as per Figure 1, while with HgCl2 the product (PhS-Hg-Cl)2·HgCl2 (6) can be formed via two routes, one involving the reaction of Hg(SPh)2 with HgCl2. Therefore, we studied the scrambling reactions between the metal thiophenolates Cd(SPh)2 (1), Pb(SPh)2 (2) and Hg(SPh)2 (3) with their acetates and chlorides, Table 2.
As can be seen from Table 2, 1 and 2 were inert towards their acetates and chlorides but 3 was reactive. Running the reaction of 3 with Hg(OAc)2 (III) in MeOH, then under 1 : 1 molar ratio in 48 h only 50% PhS-Hg-OAc (5) precipitated, while with 1 : 2 molar ratio the solid had, by 1H-NMR, the composition PhS-Hg-OAc·0.11Hg(OAc)2. Under 1 : 1 stoichiometry of 3 and III in CHCl3/MeOH 1 : 1 v/v, however, 92% PhS-Hg-OAc (5) was obtained, Table 2.
The mechanism of scrambling may involve a cyclic 6-membered transition state, cf Figure 1. The structure of 5 is polymeric having infinite (-S-Hg-S)x chains linked via acetate groups into sheets [22] and is analogous to that of MeS-Hg-OAc [2]. It is said that this may be a general feature of compounds containing Hg(II) and a thiolate, RS-, group (1 : 1 molar ratio) [2].
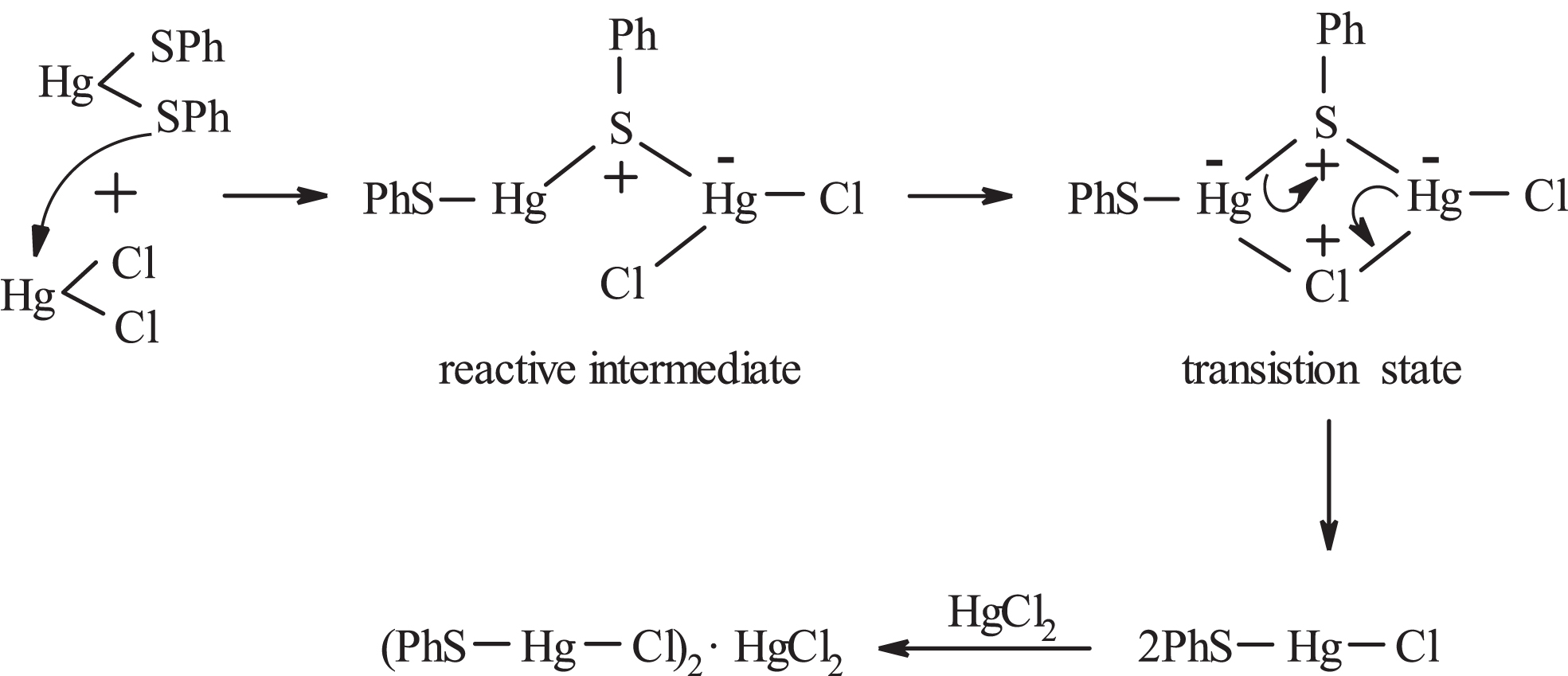
The reaction of Hg(SPh)2 (3) with HgCl2 (IV) (1 : 1, 1 : 2, 1 : 3 and 1 : 4 molar ratios) in CHCl3/Me2CO (2 : 1 v/v) gave (PhS-Hg-Cl)2·HgCl2 (6) in only 46–58% yields. The low yields may be due to a finite solubility of 6 in Me2CO used to extract any excess of HgCl2. The production of 6 should involve the formation of PhS-Hg-Cl via a 4-center transition state, Figure 4, which, then, captures one HgCl2 molecule.
Reactive intermediate and 4-centered transition state in the scrambling of the ligands between Hg(SPh)2 and HgCl2.
For ESI MS, the samples of PhS-Hg-OAc (5) and (PhS-Hg-Cl)2·HgCl2 (6) were dissolved in minimum amount of DMSO, diluted with MeOH and run at the positive mode. The spectra of both compounds were the same, the molecular ions were not detected and indicated extensive decomposition and reactions in the gas phase. Only a few ions could be identified: PhS+=O (125.13), HO-Hg+ (218.22), PhS-Hg-OH·H+ (327.18) and PhS-Hg-O-Hg+ (or its cyclic version) (528.79). Thus the ESI MS spectra cannot distinguish between 5 and 6 or between, e.g., Hg(SPh)2 and Hg(OAc)2 as separate entities. Similarly, of no diagnostic help were the IR (KBr) spectra of M(SPh)2 (M = Cd, Pb, Hg) (1)–(3) and (PhS-Hg-Cl)2·HgCl2 (6) which were identical and same as PhSSPh showing the fundamental vibrations of a mono-substituted benzene.
The reason why Hg(SPh)2 is reactive while Cd(SPh)2 and Pb(SPh)2 are not reactive towards their acetates and chlorides is presently not known. It may be due to the insolubility of Cd(SPh)2 and Pb(SPh)2 in the solvents used.
Concluding remarks
The reaction of the As(III) thiolates A, B and C with the metal acetates M(OAc)2 (M = Cd, Pb, Hg) gave (except in the case of B + Cd(OAc)2) the metal thiophenolates M(SPh)2 (1)–(3), while with HgCl2 gave (PhS-Hg-Cl)2·HgCl2 (6), Table 1. In all cases, the attacking atom was the S(II) (Figure 1), but under certain experimental conditions the As(III) of B and C can attack Hg(OAc)2 and/or PhS-Hg-OAc giving elemental mercury as an impurity. The variation in the yields of (1)–(3) and (6) may be due to the formation and to the collapse of the initially formed intermediates (i.e. the Lewis adducts). The latter probably decompose via a 6-membered (for acetates) or a 4-membered (for chlorides) transition states, Figures 1 and 4.
The claim that the reaction of various Ar-As(SPh)2 with HgCl2 in ethanol gave compounds of the type Ar-As(SPh)2·4HgCl2 [19] was not substantiated probably because mixtures of (PhS-Hg-Cl)2·HgCl2 and (Ar-AsO)x have been analyzed.
Scrambling of the substituents was observed between Hg(SPh)2 and Hg(OAc)2 or HgCl2 but not for M(SPh)2 and M(OAc)2 or MCl2 (M = Cd, Pb). The product PhS-Hg-OAc (5), Table 2, is stable in the absence but not in the presence of PhS- groups, Figure 1, while the initially formed PhS-Hg-Cl captures one HgCl2 molecule to give the insoluble (PhS-Hg-Cl)2·HgCl2 (6) (a class A adduct of Evans et al.).
The IR (KBr) spectra of M(SPh)2 (1)–(3) and (PhS-Hg-Cl)2·HgCl2 (6) are identical and similar to that of PhSSPh. The ESI mass spectra of 5 and 6 revealed absence of molecular ions and extensive fragmentations that cannot be easily rationalized.
While the object of the present work was the investigation of the reactions of As(III) thiolates with heavy metal cations, the results point to simpler and more general reactions to obtain new Hg(II) adducts avoiding the use of As(III) compounds.
Thus, instead of using Equation (3), the Equation (8) can be used to prepare RS-Hg-OAc with a wide variety of RS- groups. An arsenic-free preparation of RS-Hg-OAc used two Hg(II) compounds, Hg(SR)2 and Hg(OAc)2, under 1 : 1 stoichiometry [2].
An even wider spectrum of mixed Hg(II) compounds can be made based on Equation (9) which requires only the preparation of Hg(II) carboxylates e.g. from HgO and a carboxylic acid.
Also, instead of using Equations (4) and (5), the arsenic-free reaction of Equation (10) can be used for the preparation of (RS-Hg-Cl)2·HgCl2 with a variety of RS- instead of PhS- groups. It should be noted that the reaction of RSH and HgX2 under 1 : 1 stoichiometry produces RS-Hg-X [2].
Footnotes
Acknowledgments
We thank Professor R. Winpenny and Dr E. Timco (University of Manchester, England) for the elemental analyses reported herein.
References
1.
CantyA.J., KishimotoR.Mercury(II) and organomercury(II) complexes of thiols and dithiols, including British Anti-ite Lewis , Inorg Chim Acta24 (1977), 109–122.
2.
CantyA.J., KishimotoR, TysonR.K., Synthesis and spectroscopic studies of RSHgII complexes, including complexes of ButSHgX (=Cl, Br) and RSHgO2CMe (=Me, Et) with pyridine, Aust J Chem31 (1978), 671–676.
3.
CantyA.J., RastonC.L., WhiteA.H., Crystal and molecular structure of polymeric MeSHgO2CMe and MeSHgO2CMe,C5H5N and the tetranuclear complex (ButS)4Cl4Hg4(C5H5N)2, , Aust J Chem31 (1978), 677–684.
4.
ChaudhryS.C., MahajanR.K., BhattS.S., SharmaN.A new method of synthesis of arsenic trithiophenoxide, , Indian J Chem31A (1992), 279–280.
5.
DanceI.G., GarbuttR.G., CraigD.C., ScudderM.L.The different nonmolecular polyadamantanoid crystal structures of Cd(SPh)2 and Cd(SC6H4Me-4)2. Analogies with microporous aluminosilicate frameworks, , Inorg Chem26 (1987), 4057–4064.
6.
DavisA.R., MurphyC.J., PlaneR.A.Triphenylphosphine and triphenylarsine complexes of mercury(II) thiocyanate, nitrate, and perchlorate, , Inorg Chem9 (1970), 423–425.
7.
DurrantP.J., DurrantB. Introduction to Advanced Inorganic Chemistry, Longmans, London, 1962, p. 748.
8.
DykeW.J.C., DaviesG., JonesW.J. Some aliphatic compounds of arsenic, J Chem Soc (1931), 185–188.
9.
EvansR.C., MannF.G., PeiserH.S., PurdieD. The composition of complex metallic salts. Part 310 XI. The structure of the tertiary phosphine and arsine derivatives of cadmium and mercuric halides, J Chem Soc (1940), 1209–1230.
10.
HaikouM.N., IoannouP.V.The autoxidation of triaryl trithioarsenites, (ArS)3As: Evidence for binding and activation of triplet dioxygen by arsenic(III), Sulfur, and Silicon181 (2006), 363–376.
11.
IoannouP.V.The use of bismuth nitrate pentahydrate, Bi(NO3)3·5H2O, and bismuth subnitrate monohydrate, BiO(NO3)·H2O, for the preparation of tris(arylthio)bismuthines, , Main Group Chem10 (2011), 255–264.
12.
IoannouP.V.Trithioarsenites (RS)3As], dithioarsonites [R-As(SR′)2] and thioarsinites [R2As-SR′]: Preparations, chemical, biochemical and biological properties, Main Group Chem21 (2022), 585–610.
13.
IoannouP.V., AfroudakisP.A., SiskosM.G.Preparation of 2-picolylarsonic acid and its reductive cleavage by ascorbic acid/iodine and by thiophenol, , Phosphorus, Sulfur, and Silicon177 (2002), 2773–2783.
14.
KamaiG.Synthesis and properties of esters of trivalent arsenic, Uchenye Zapiski Kazan. Gosudarst Univ115 (1955), 43–45; Chem Abstr 53 (1959), 1205e.
15.
KamaiG., ChadaevaN.A.Action of halides and anhydride of acetic acid on glycol esters of phenylarsonous acid. Dokl Akad Nauk S.S.S.R.115 (1955), 309–311;Chem Abstr 51 (1957), 1876h.
16.
LangE.S., DiasM.M., dos SantosS.S., Vázquez-LópezE.M., AbramU.Synthesis and structure of the clusters [Hg2(μ-SePh)2(SePh)2(PPh3)2] and [Hg3Br3(μ-SePh)3]·2DMSO, , Z Anorg Allg Chem630 (2004), 462–465.
17.
LongL.H., EmeleusH.J., BriscoeH.V.A. The difluoroarsines, J Chem Soc (1946), 1123–1126.
18.
NesmejanowA.N., KozeschkowK.ADie Reaktionen von arsen-, antimon-, zinn- und blei-organischen Verbindungen mit Quecksilberchlorid im neutralen und alkalischen Medium, , Chem Ber67 (1934), 317–324.
19.
SchusterM.G.Contribution à l’ étude de quelques arylarsinites de thiophenol et de leurs complexes mercuriques, , J Pharm Chim17 (1933), 331–334.
20.
ServesS.V., CharalambidisY.C., SotiropoulosD.N., IoannouP.V.Reactions of arsenic(III) oxide, arsenous and arsenic acids with thiols, , Phosphorus, Sulfur, and Silicon105 (1995), 109–116.
21.
ShawR.A., WoodsM. Preparation and some properties of lead thiolates, J Chem Soc A (1971), 1569–1571.
22.
StålhandskeC., ZintlF.Structure of acetato(benzenethiolato)mercury(II), Acta CrystC42 (1986), 1731–1733.
23.
TerzisA., IoannouP.V.On the reaction of dithioasonites L-As(SPh)2 (=Ar, R), with octasulfur in the presence of triethylamine as an activator. The crystal structure of the sesquisulfide (2-O2N-C6H4-As)2S3, Z Anorg Allg Chem630 (2004), 278–285.
24.
TsivgoulisG.M., SotiropoulosD.N., IoannouP.V.Decomposition of alkylarsonic acid by acylating agents, Phosphorus, Sulfur, and Silicon55 (1991), 165–168.

 (
(