
Abstract
The adsorption of the dibenzothiophene (DBT) molecule upon the boron nitride nanosheet (BNNS) was discussed using the DFT method by M062X/6-311 + G* level of theory in the water solvent. The results of thermochemical parameters display the interaction of the DBT with BNNS is a spontaneous and exothermic process. The UV/Vis absorption analysis was carried out to predict the changes that occurred during the adsorption of the DBT upon the BNNS. Based on the FMO analysis, the value of the energy gap (Eg) of the BNNS reduced after the interaction of the DBT with the BNNS. The negative value of ΔN (-0.0048) of the DBT@BNNS complex confirms the charge transfer from DBT to the BNNS which is inconsistent with the results of the NBO analysis. QTAIM analysis displays an electrostatic interaction between BNNS and DBT. According to the results of NICS calculations, after the interaction of DBT with BNNS, all three rings A, B, and C of DBT have become more aromatic and stable in the presence of the nanosheet magnetic field. We hope that our findings can be used for modeling and designing a suitable adsorbed for the adsorptive desulfurization process.
Introduction
The desulfurization process from fuels is a significant topic for decreasing sulfur oxide compounds in the atmosphere [1]. Sulfur oxide compounds react with drops of water and produce acid rain which causes damage to the environment and corrosion of equipment and devices [2]. The release of sulfur compounds in the atmosphere also harms human health [3]. Hydrogen, carbon, and sulfur are the most abundant elements in diesel fuel [4]. To increase the quality of fuels, sulfur compounds are removed from crude oil. One of the important techniques for the removal of organic sulfur compounds from oil is the adsorptive desulfurization technique using the physicochemical adsorption method [5–7]. Boron nitride nanosheets with exclusive properties including high thermal stability and conductivity, wide surface, low density, electrical insulation, and resistance to oxidation have been used in the adsorption process [8–13]. Because of these attractive properties, researchers have paid attention to boron nitride nanosheets as suitable adsorbents for the removing of sulfur compounds from fuels [14–17]. Based on the theoretical calculations, the strong π-π interactions occur between boron nitride nanosheets and thiophenic compounds; therefore it is very suitable in the adsorptive desulfurization process [18]. Li and co-workers have studied the effects of O-doping into the boron nitride nanosheet in the desulfurization process using the DFT method [19]. They also have reported comparative research on the halogen-doped (X = F, Cl, Br, I) into the BNNS in the adsorptive desulfurization process [20, 21]. In another study, Li and co-workers have reported the synthesis of boron nitride mesoporous nanowires with doped oxygen atoms for the remarkable adsorption desulfurization performance from fuels [22]. The obtained BN mesoporous nanowires have displayed good stability and outstanding adsorptive desulfurization activity for DBT in comparison of commercial BN and graphene-like BN. According to the experimental report of Zhu and co-workers, the hexagonal boron nitride nanosheets stabilized platinum nanoparticles (Pt/h-BNNS) active adsorbent showed excellent desulfurization performance with the existence of other interferents such as aromatic hydrocarbons or olefins [23]. Lin and co-workers have investigated the catalytic activity of Metal-doped BNNS in CO Oxidation by theoretical calculations [24]. Lu and co-workers have discussed the single Ag atom protected on the BNNS as a catalyst in the CO oxidation process to CO2 by theoretical calculations [25]. Liu et al. have studied the attached single Ag atom on faulty BNNS for increasing adsorptive desulfurization through S-Ag bonds [26].
In the current study, the adsorption conduct of the dibenzothiophene (DBT) molecule on the pristine BNNS is presented by DFT calculations. Actually, we have studied the effect of nanosheet on the electronic behavior of the DBT molecule in desulfurization process. The geometric and thermochemical parameters and various analyses including the frontier molecular orbitals (FMO) analysis [27], natural bond orbital (NBO) analysis [28], charge difference (ΔN) [29], Nucleus-Independent Chemical Shifts (NICS) analysis [30], and UV/Vis absorption analyses were discussed. Also, QTAIM analysis has been carried out to study the nature of the bond and interaction between nanosheet (BNNS) and DBT. We believe that the obtained results can give a good viewpoint for the application of BNNS as an adsorbent platform in the process of desulfurization of fuels and also modeling other platforms.
Computational methods
The calculations of adsorption of the dibenzothiophene (DBT) over boron nitride nanosheet (BNNS) have been predicted using the M062X/6-311 + G* level. All calculations were carried out by Gaussian 09 software [31] in the water solvent. The Polarized Continuum Model (PCM) has been used for the solvent effect [32]. The adsorption energies (Eads) [33] of the DBT upon BNNS were calculated by the Equation (1):
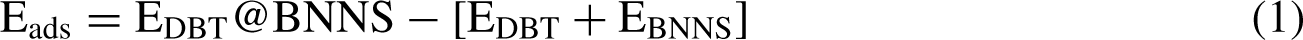




Where the μA and μB are the chemical potentials of the DBT and BNNS respectively, and the ηA and ηB are the global hardness of the DBT and BNNS respectively. GaussView 05 software [35] was used to obtain the pictures of optimized compounds and molecular orbitals. Total electronic densities of states (DOSs) graphs were obtained by the GaussSum 2.2 software [36]. NBO analysis was done to identify the donor-acceptor transitions of the DBT@BNNS [37]. The TD-DFT method [38] was used to perform the UV-Vis absorption analysis. The Quantum theory of atoms in molecules (QTAIM) analysis was performed by MultiWFN 3.1 software [39].
Optimized molecules
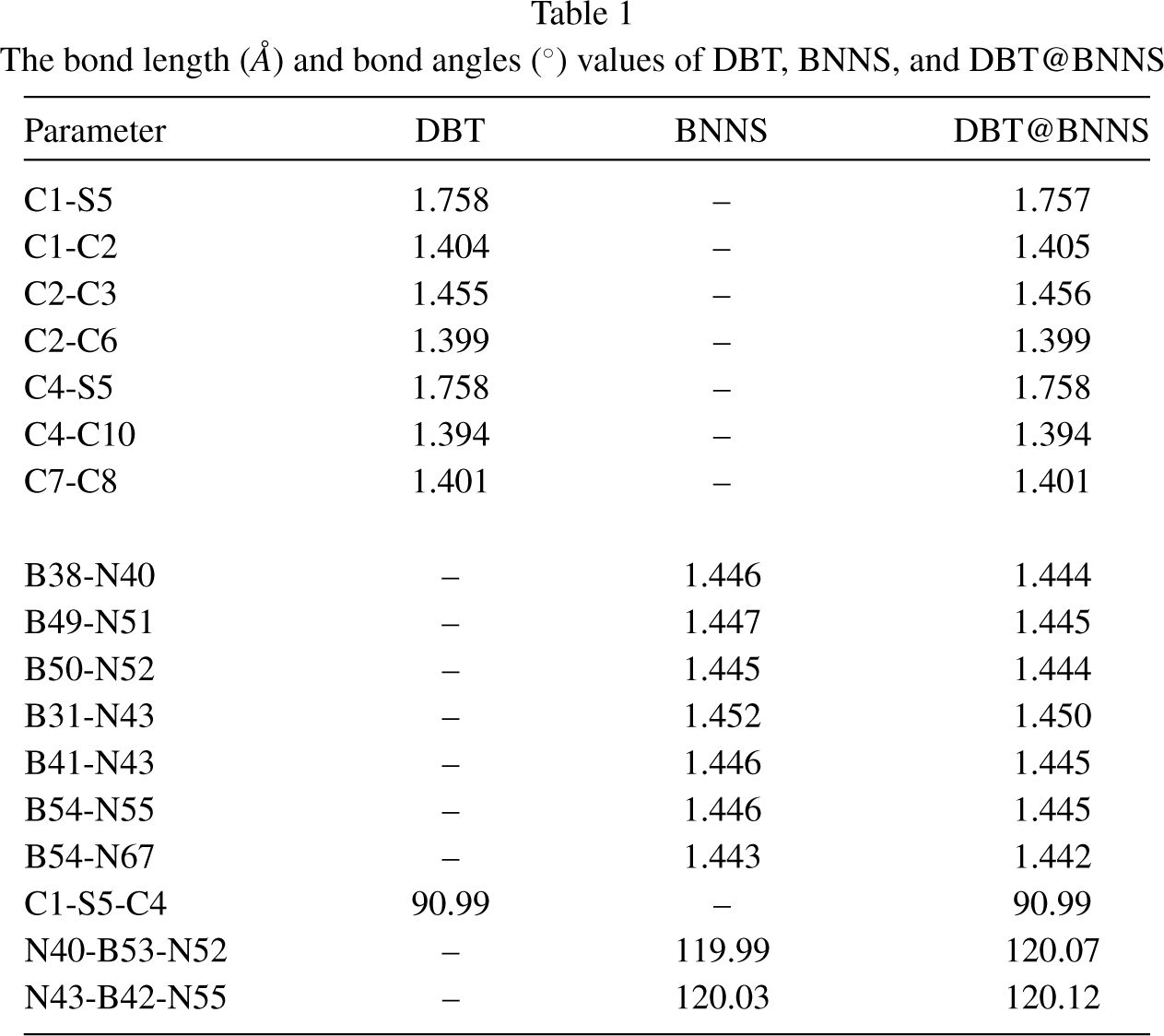
The DBT, BNNS, and DBT@BNNS complex have been optimized using the DFT method. Figure 1 represents the optimized compounds. The geometric parameters of the DBT, free BNNS, and DBT@BNNS complex were reported in Table 1. The value of C1– S5 bond length in free DBT molecule is 1.758 Å, whereas after the interaction of DBT and BNNS decreases to 1.757 Å. The length of B– N bonds in the DBT@BNNS complex is shorter than free BNNS. The values of the C1-S5-C4 bond angle in both free DBT and DBT@BNNS complex are 90.99°; therefore this bond angle does not change after the adsorption process. The N40-B53-N52 and N43-B42-N55 bond angles in the free BNNS are 119.99° and 120.03°, while after the adsorption process increase to 120.07° and 120.12°, respectively. The change of the value of the parameters value demonstrates the interaction of the DBT molecule with the BNNS.
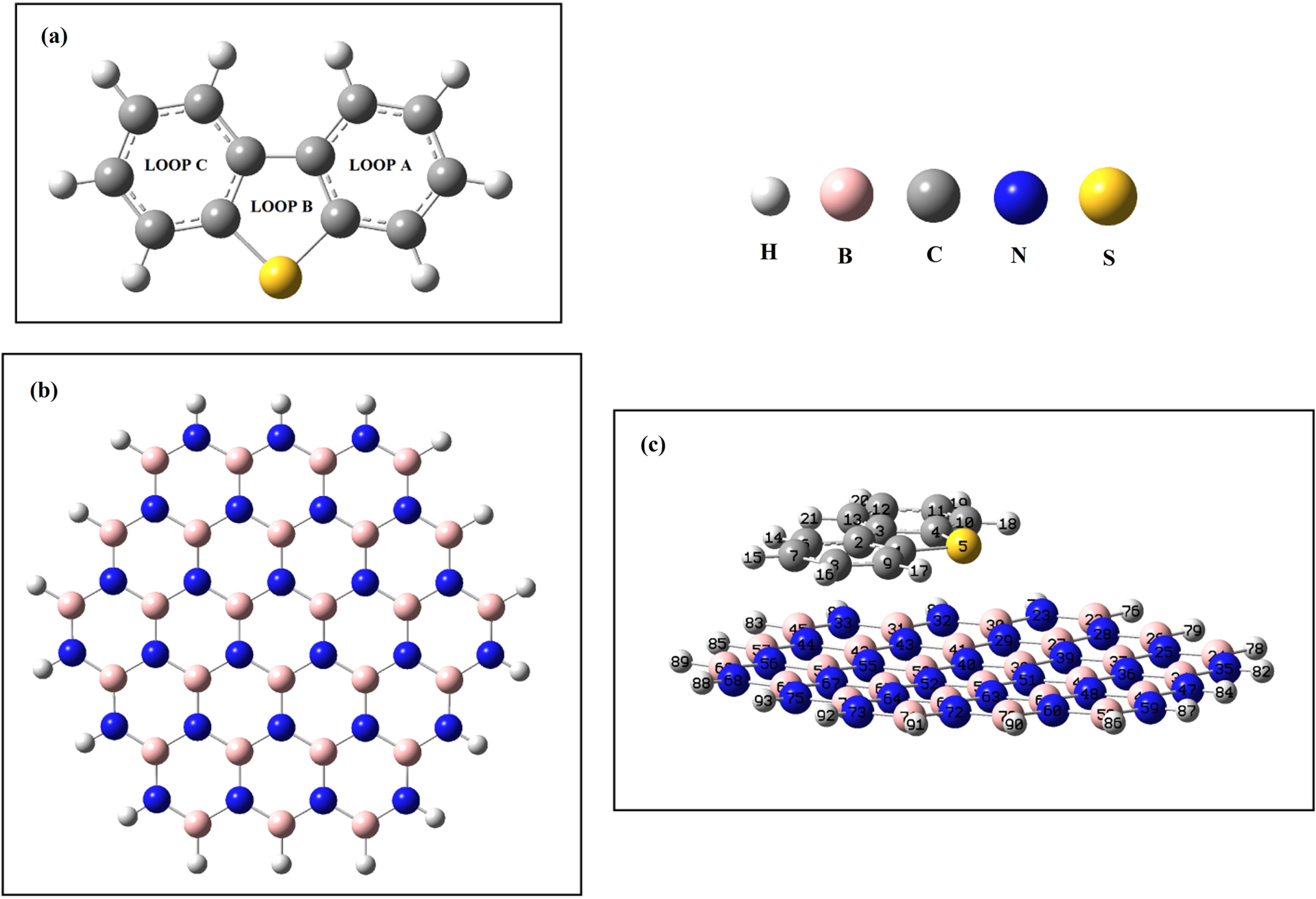
The optimized DBT, BNNS, and DBT@BNNS complex.
The bond length (Å) and bond angles (°) values of DBT, BNNS, and DBT@BNNS
The calculated results of thermochemical parameters such as including the sum of electronic and thermal energies (E + T), sum of electronic and thermal enthalpies (E + H), sum of electronic and thermal free energies (E + G), and sum of electronic and zero-point energies (ZPE) of the the DBT, BNNS, and DBT@BNNS complex were summarized in Table 2. Based on data, the adsorption of DBT over the BNNS reduces the energy values and reactivity reduces with the adsorption of DBT over the BNNS and the DBT@BNNS becomes more stable in comparison to free BNNS. In the adsorption reaction:
The values of thermodynamic parameters of the DBT, BNNS, and DBT adsorbed upon the BNNS
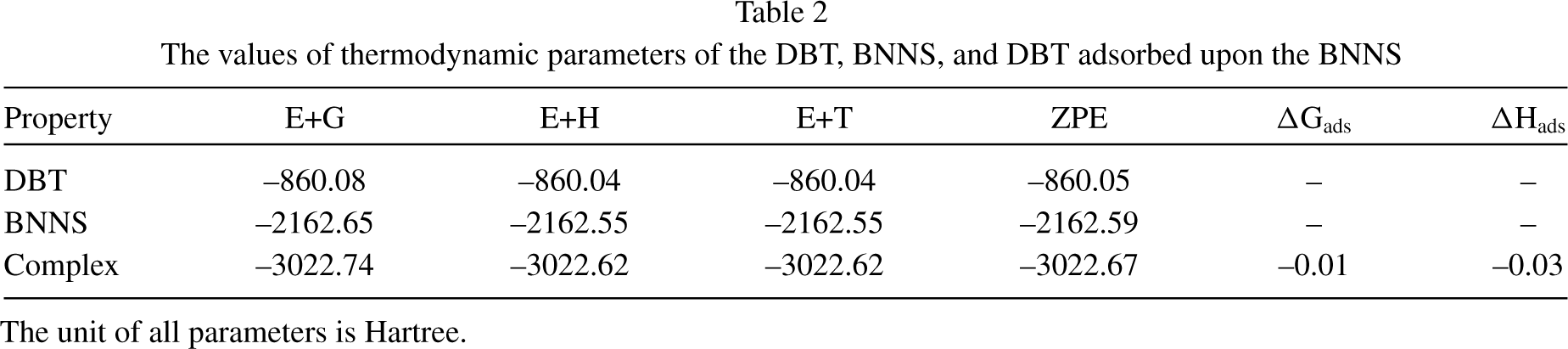
The unit of all parameters is Hartree.
DBT + BNNS ⟶ DBT@BNNS
ΔGads=GDBT @ BNNS– [GDBT+GBNNS]
ΔHads=HDBT @ BNNS– [HDBT+HBNNS]
the change Gibbbs free energy (ΔGads) is the thermodynamic quantity that shows whether a reaction is spontaneous or not. A process is thermodynamically possible and spontaneous when the ΔGads is negative (ΔGads<0). The positive value of ΔGads (ΔGads>0) shows the process is non-spontaneous and cannot happen. The change Entaply (ΔHads) includes changes in kinetic and potential energies at constant pressure while the reaction is taking place. We have performed the calculations at constant pressure (1 atm). A reaction is considered energetically feasible when the energy of the system decreases as a result. In the other words, make the system more stable. In this case, the ΔHads will be negative (ΔHads<0). Besides, a reaction that has a ΔHads>0 is an endothermic process, while ΔHads<0 shows an exothermic process. The negative value of ΔGads and ΔHads values shows that the interaction of DBT with BNNS is spontaneous and exothermic, respectively.
The calculated results of electronic properties before and after the adsorption process were summarized in Table 3. The formation of polar bonds between the DBT and BNNS leads to an increase in the value of dipole moment (DM) of the DBT@BNNS (1.823 Debye) in comparison to free BNNS (0.020 Debye) and this change approves a process of charge transfer between the DBT and BNNS. The negative value of Eads (-0.73 eV) displays the exothermic nature of the adsorption process. The pictures of HOMO and LUMO orbitals of the DBT, BNNS, and DBT@BNNS complex are observed in Fig. 2. The HOMO orbital of the nanosheet is located on the nitrogen atoms, whereas the LUMO orbital is located on the boron atoms on the edge of the nanosheet. The HOMO orbital of the DBT is located on the rings A and C, and sulfur atom of ring B, whereas the LUMO orbital is located on the rings A and C. In the DBT@BNNS complex, the HOMO and LUMO orbitals focused on the DBT section. The values of EHOMO, ELUMO, and energy gap (Eg) of DBT, BNNS, and DBT@BNNS are shown in Table 3. The value of Eg of the free BNNS is 13.99 eV; whereas the value Eg and conductivity of the DBT@BNNS are reduced (10.49 eV). DOS plans also confirm the adsorption of DBT over the BNNS and it also shows The changes in the value of Eg and electron density during the interaction of the DBT with BNNS also are approved with DOS plans (Fig. 3).
The electronic properties of the DBT, BNNS, and the DBT@BNNS
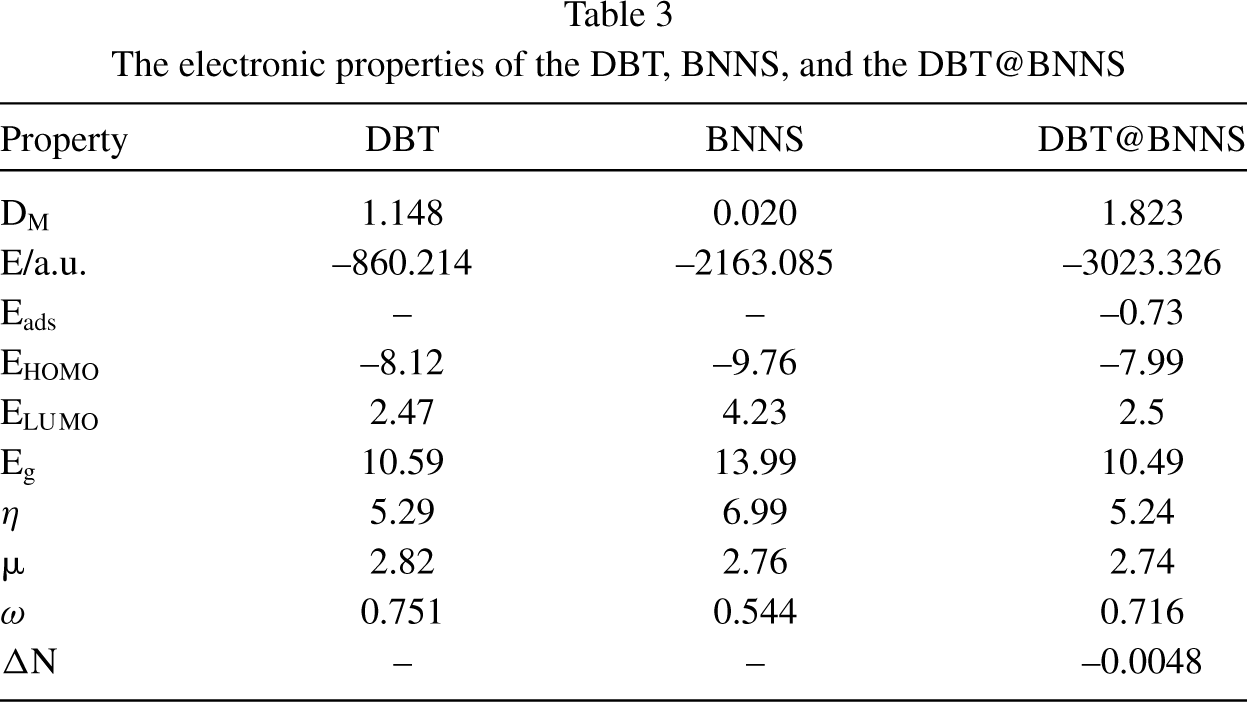
The electronic properties of the DBT, BNNS, and the DBT@BNNS
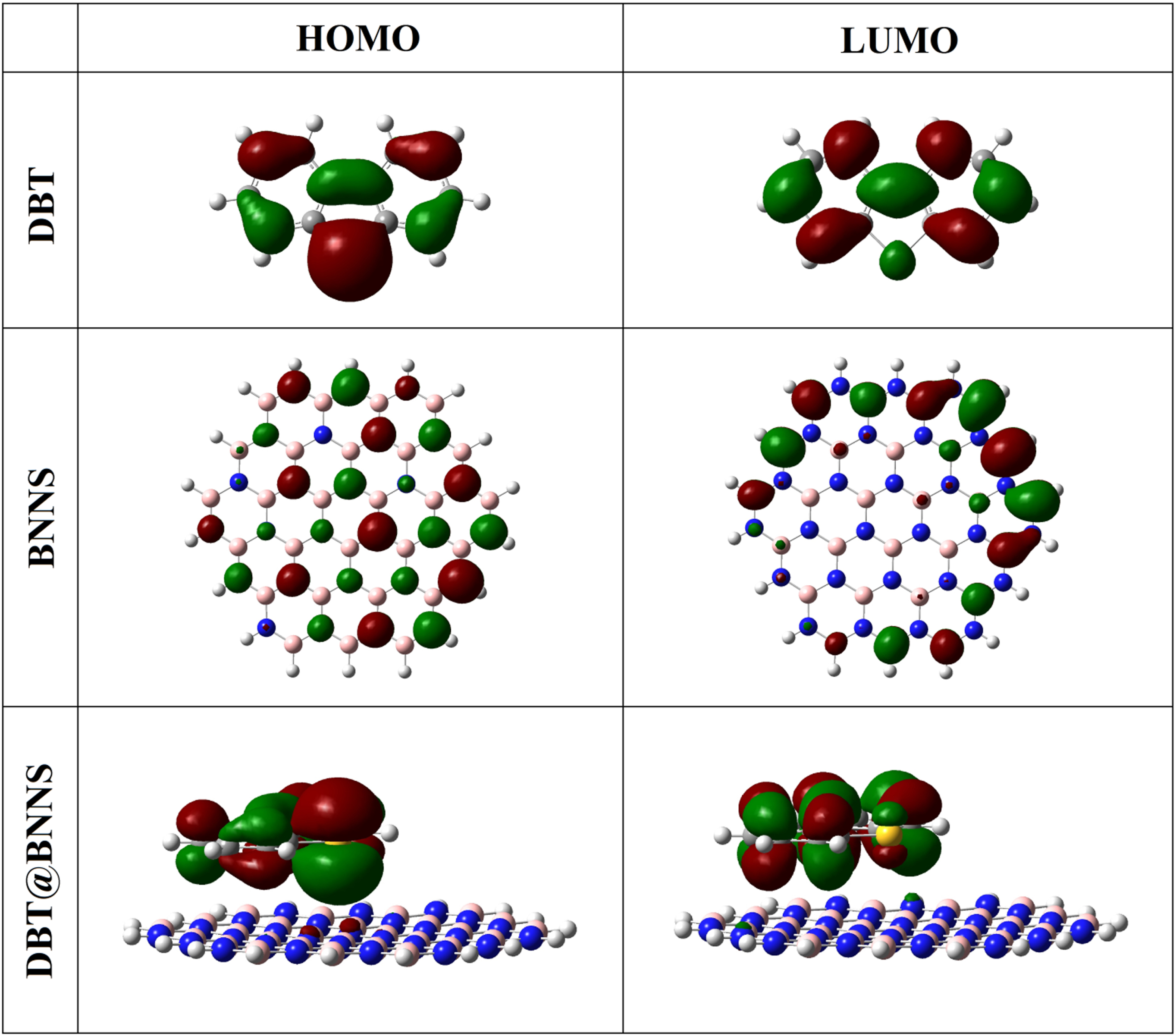
The pictures of HOMO and LUMO orbitals of the DBT, BNNS, and DBT@BNNS.
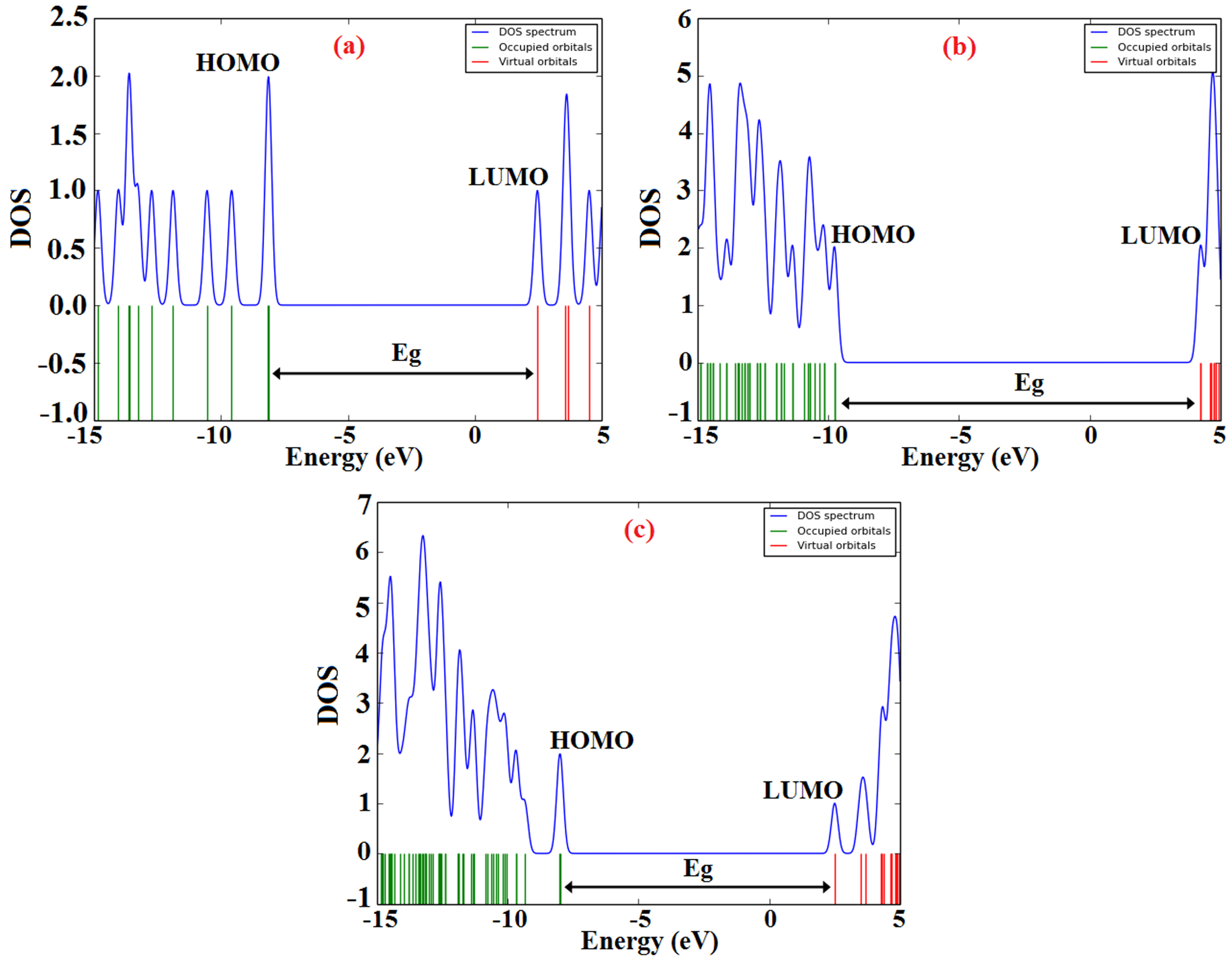
DOS graphs of the DBT, BNNS, and DBT@BNNS complex.
The ΔN value of the DBT@BNNS was calculated and reported in Table 3. The ΔN>0 value shows the charge transfer from BNNS to the DBT and the ΔN<0 value shows the charge transfer from DBT to the BNNS. The negative value of ΔN of the DBT@BNNS complex confirms the charge transfer from the DBT to the BNNT.
An aromatic ring current is an effect is observed in aromatic compounds. If a magnetic field is directed perpendicular to the plane of the aromatic molecule, a ring current is induced in the delocalized π electrons of the aromatic ring [40]. This is a direct consequence of Ampère’s law; since the electrons involved are free to circulate, rather than being localized in bonds as they would be in most non-aromatic molecules, they respond much more strongly to the magnetic field. Aromatic ring currents are relevant to NMR spectroscopy, as they dramatically influence the chemical shifts of 1 H nuclei in aromatic molecules [41]. The effect helps distinguish these nuclear environments and is therefore of great use in molecular structure determination. The nucleus-independent chemical shift (NICS) is a computational method that calculates the absolute magnetic shielding at the center of a ring. The values are reported with a reversed sign to make them compatible with the chemical shift conventions of NMR spectroscopy [42]. In this method, negative NICS values indicate aromaticity and positive values antiaromaticity. NMR calculations were performed using the GIAO-M062X/6-311 + G* level and values of magnetic shielding tensor (σisotropic) for the atoms (Bq) at the center of rings of the free DBT and the DBT in DBT@BNNS complex were calculated and reported in Table 4. The negative NICS values indicate aromaticity in the rings [43] of the free DBT molecule and the adsorbed DBT upon the surface of BNNS. The NICS value can be calculated at the ring center (NICS (0)), at 1Å above the ring center (NICS (1)), or 1Å below the ring center (NICS (-1)). Generally, NICS > 0 indicates a paratropic ring current, and NICS < 0 shows a diatropic ring current. Also, NICS (0) belongs to the non-aromatic species. Based on the results of the NICS analysis, the NICS values at the B ring of the free DBT at the ring center, 1Å above the ring center, and 1Å below the ring center are -4.2665, -6.4513, and -6.4475 ppm respectively, which displays the maximum ring current is at the 1Å above the ring center with the value of -6.4513 ppm. The A and C rings of the free DBT have identical NICS values. NICS values at the center of the rings, 1Å above the center of the rings, and 1Å below the center of the rings including -7.2491, -10.1371, and -10.1358 ppm, respectively; therefore maximum ring current is 1Å above the A and C rings center with the value of -10.1371 ppm. After interaction of DBT with the BNNS, the NICS values at the B ring of the DBT in the DBT@BNNS complex at the ring center, 1Å above the ring center, and 1Å below the ring center are -7.9611, -7.3186, and -7.0063 ppm, respectively; that indicates the maximum ring current is at the ring center with the value of -7.9611 ppm. At the A and C rings of the DBT in the DBT@BNNS complex, the maximum ring current is 1Å above the ring center with values of -11.3355 and -11.3516 ppm, respectively. Thus after the interaction of DBT with BNNS, all three rings A, B, and C of DBT have become more aromatic and stable in the presence of the nanosheet magnetic field. Also after the adsorption process, the aromaticity A and C rings are not identical and the C ring has more aromaticity rather than the A ring.
NICS values of the FREE DBT and adsorbed DBT upon the BNNS

NICS values of the FREE DBT and adsorbed DBT upon the BNNS
NBO analysis was done to identify the donor-acceptor transitions of the DBT@BNNS. A molecule with higher resonance energy (E (2) ) is the more stable. The results of the NBO analysis such as the electron donor orbitals, acceptor orbitals and the interacting stabilization energy (E (2) ) of the DBT@BNNS are represented in Table 5. The charge transfer from the LP(2)S5 of the DBT to the BD*(2)B38-N51 and BD*(2)N40-B53 antibonding orbitals of the BNNS were detected as the LP(2)S5⟶ BD*(2)B38-N51 and LP(2)S5⟶ BD*(2)N40-B53 transitions with E (2) 1.05 and 1.23 kcal/mol, respectively; therefore, the charge transfer occurs from the DBT to the BNNS. The lowest value E (2) is observed for the BD(1)C11-H19⟶BD*(2)B31-N33 transition. The charge transfer also is done from BNNS to the DBT molecule. For example, the BD(2)N29-B41 bonding orbital of the BNNS interacts with the LP(1)C10 and of DBT as the BD(2)N29-B41⟶LP(1)C10 transition with E (2) 0.81 kcal/mol. The BD(2)B31-N33⟶LP*(1)C12, BD(2)N40-B53⟶LP(1)C4, and BD(2)B54-N55⟶LP(1)C2 transitions are the other interactions of the DBT@BNNS, from the BNNS to the DBT, with E (2) 0.65, 0.63, and 0.76 kcal/mol, respectively.
The important transitions of NBO analysis of the DBT@BNNS complex

The important transitions of NBO analysis of the DBT@BNNS complex
Also, the results of NBO analysis of the free DBT molecule, the DBT in the complex, and some of the electronic transitions related to the thiophene ring have been reported in Table 6. Based on the results, the highest E (2) of the free DBT and DBT in complex is observed for the BD(1)C8-C9⟶ BD*(1)C1-S5 about 5.62 and 5.63 kcal/mol respectively. Also, the lowest E (2) of the free DBT and DBT in complex is observed for the BD(1)C4-C10⟶ BD*(1)C4-S5 about 0.52 and 0.59 kcal/mol respectively. Other energies can be seen in Table 6. When the DBT molecule and BNNS are affected by each other’s magnetic field, the E (2) of the most electronic transfers in the thiophene ring of the complex increases compared to the free DBT molecule. Beside, the E (2) of the BD(1)C1-S5⟶ BD*(1)C3-C4 and BD(1)C4-S5⟶ BD*(1)C1-C2 transitions in the thiophene ring of the complex decreases compared to the free DBT, that show theses molecular orbitals in the complex are unstable compared to the free DBT and desulfurization can be done more easily.
Second order perturbation theory analysis of fock matrix in NBO analysis
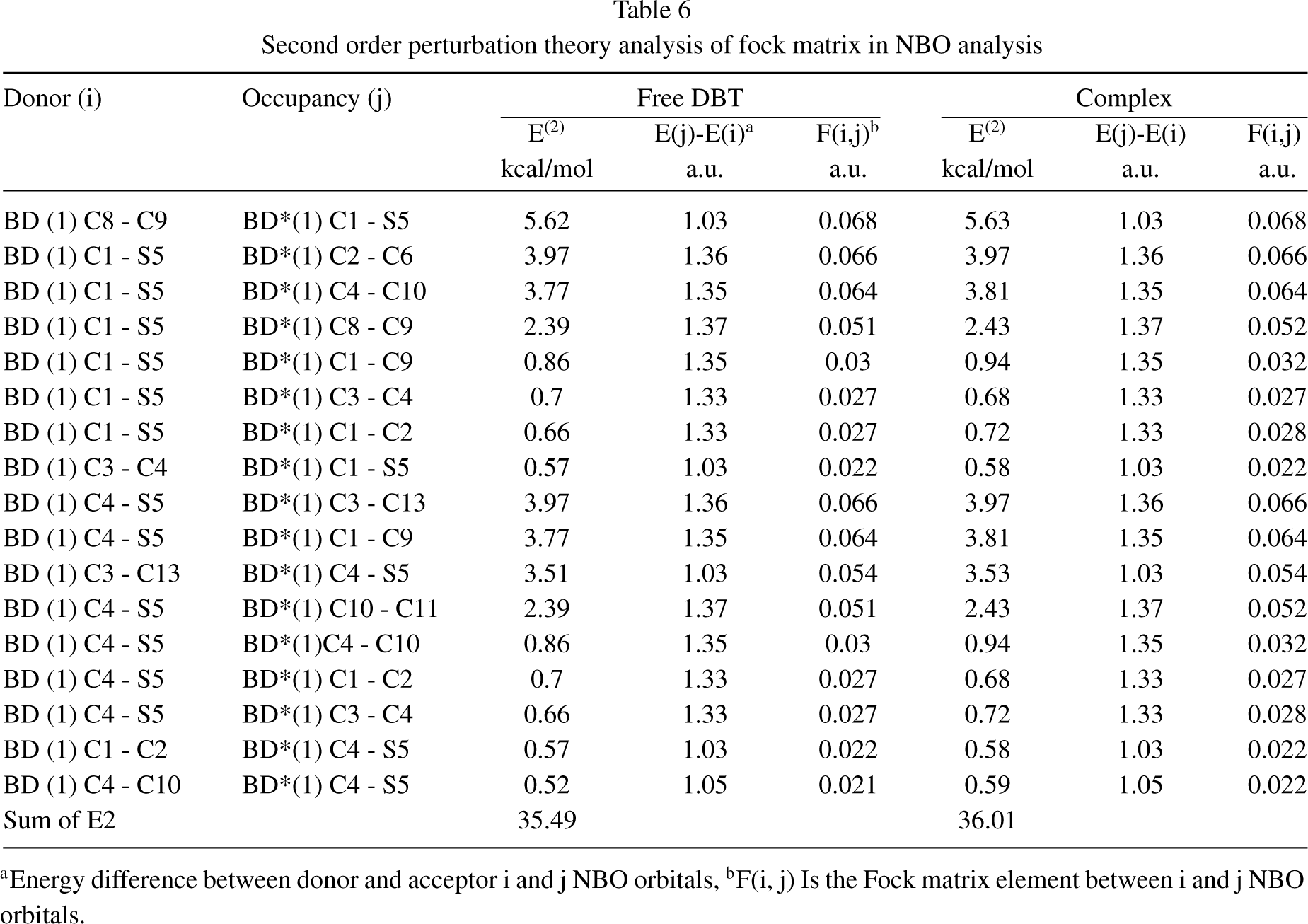
aEnergy difference between donor and acceptor i and j NBO orbitals, bF(i, j) Is the Fock matrix element between i and j NBO orbitals.
The total E (2) in the thiophene ring of the complex is about 36.01 kcal/mol, whereas the total E (2) in the thiophene ring of the free DBT is about 35.49 kcal/mol. The increase in the total E (2) shows that the reactivity and aromaticity of the DBT molecule in the presence of BNNS and confirms that desulfurization is easier. The results of the NICS analysis is also a confirmation of this fact, because the NICS values with aromaticity in the Loop B (thiophene ring) of the complex increased compared to the free DBT.
To study the nature of the bond and interaction between nanosheet (BNNS) and DBT, topological parameters in the bond critical points (BCPs) [44] such as electron density (ρ
BCP
), Laplacian of electron density (∇2ρ
BCP
), kinetic energy density (G
BCP
), potential energy density (V
BCP
), and total energy density (H
BCP
) were calculated by QTAIM analysis, and the results are represented in Table 7. The ∇2ρ
BCP
> 0 and H
BCP
> 0 values display an electrostatic interaction between BNNS and DBT that is related to weak or medium bonds weak ionic bonds, ionic-hydrogen bonds, and van der Waals bonds. The ∇2ρ
BCP
> 0 value of all bonds is positive (∇2ρ
BCP
> 0) and the G
BCP
(or G
r
) in the bond critical points is greater than potential energy density (V(r)); therefore, the electron density is focused toward the nucleus (not toward critical points) and represent weak bonds between BNNS and DBT. The high value of ρ and the more positive value of ∇2ρ(r) indicates a strong bond [45]. The highest electron density is in critical points between the S5 atom and the N40. Thus, the strongest interaction in the DBT@BNNS complex with the largest values of ρ
BCP
and ∇2ρ
BCP
is observed between S5 and N40. At the critical points of all bonds, the
Topologic parameters of the bond critical points in interaction of the DBT with the BNNS
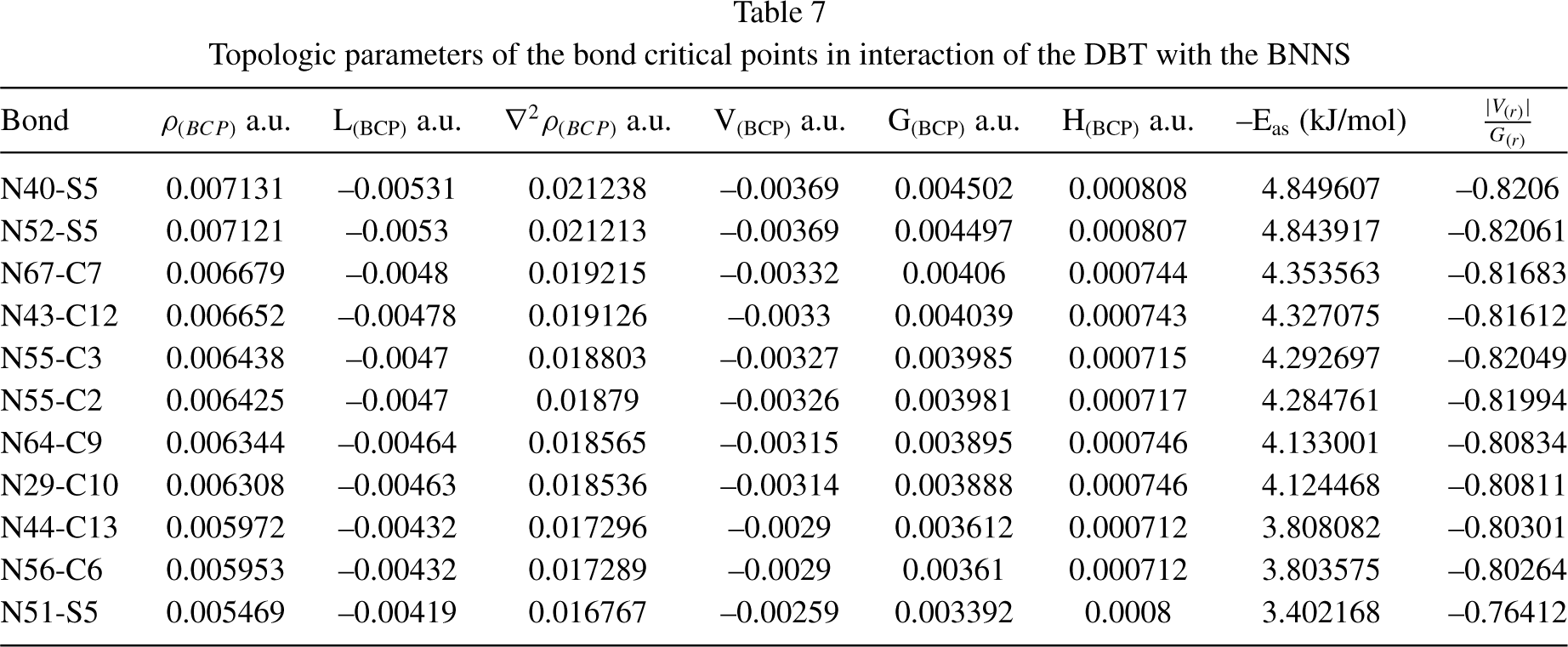
Topologic parameters of the bond critical points in interaction of the DBT with the BNNS
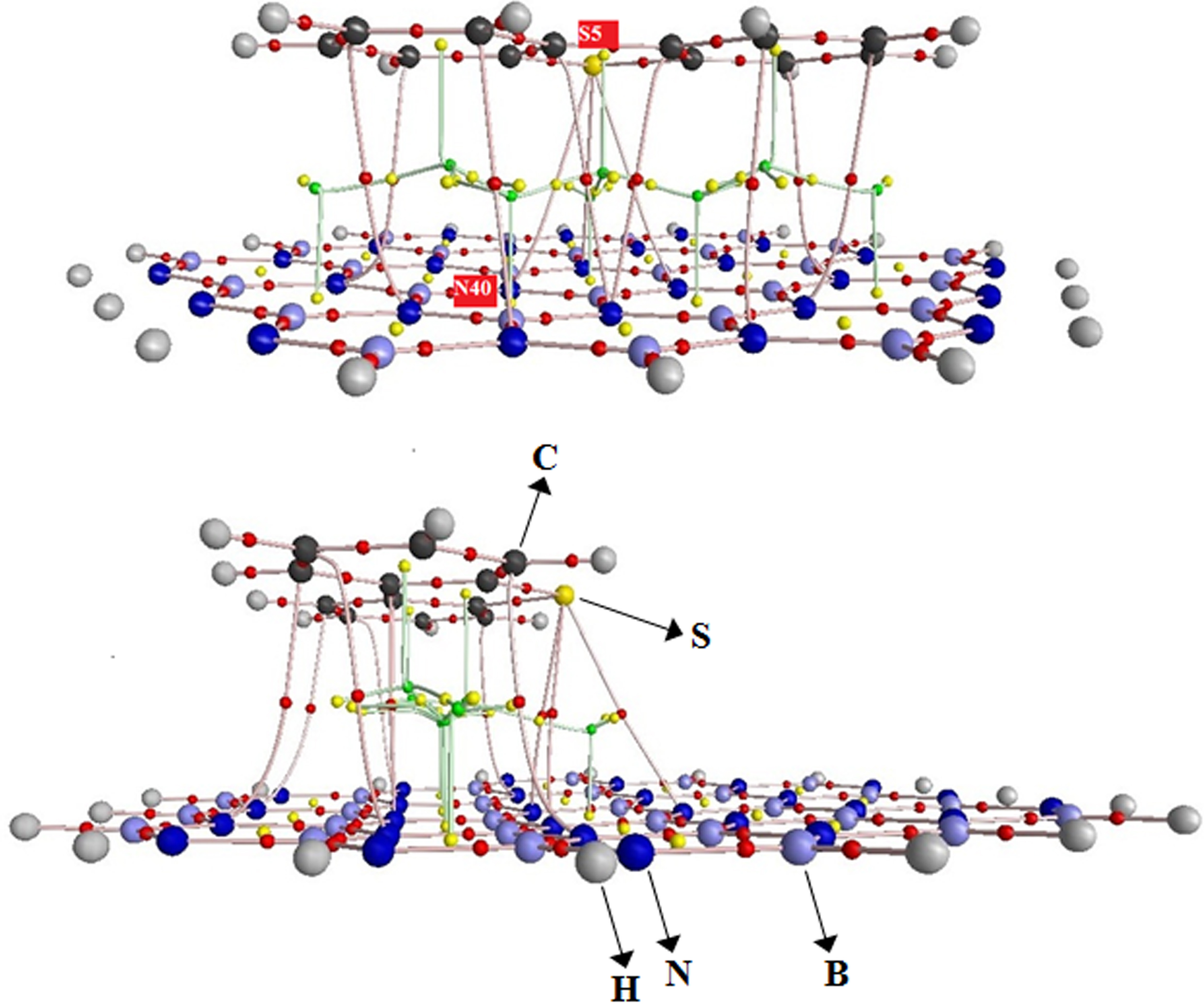
Molecular graph of DBT@BNNS complex in water solution. Small red balls: bond critical points; Small yellow balls: ring critical points.
The theoretical UV-Vis absorption of the DBT molecule, BNNS, and DBT@BNNS was obtained by the TD-DFT method and the important results are summarized in Table 8. The λmax of DBT molecule, BNNS, and DBT@BNNS is observed in the UV range. The value of λmax of the free BNNS is observed at 133.37 nm which is related to the excited state of S0⟶S14 and H-3⟶L+3 (5%) transition. The interaction of DBT with nanosheet leads to the increase of the λmax of the DBT@BNNS to 187.14 nm (redshift) which is related to the excited state of S0⟶S6. The H⟶L+2 (55%) transition is important transition in λmax=187.14. The value of λmax of the DBT molecule is observed at 184.46 nm which is related to the excited state of S0⟶S6 and H⟶L+2 (58%) transition. After adsorption process, the λmax increase to 187.14 nm (redshift). The The UV spectra of investigated compounds are presented in Fig. 5.
Significant results of UV-Vis absorption of DBT, BNNS, and DBT@BNNS
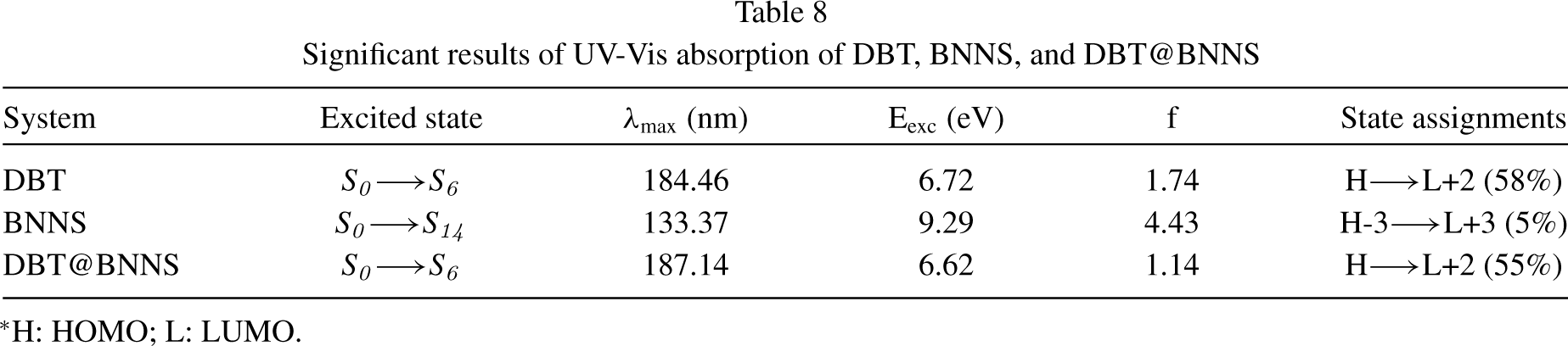
Significant results of UV-Vis absorption of DBT, BNNS, and DBT@BNNS
*H: HOMO; L: LUMO.
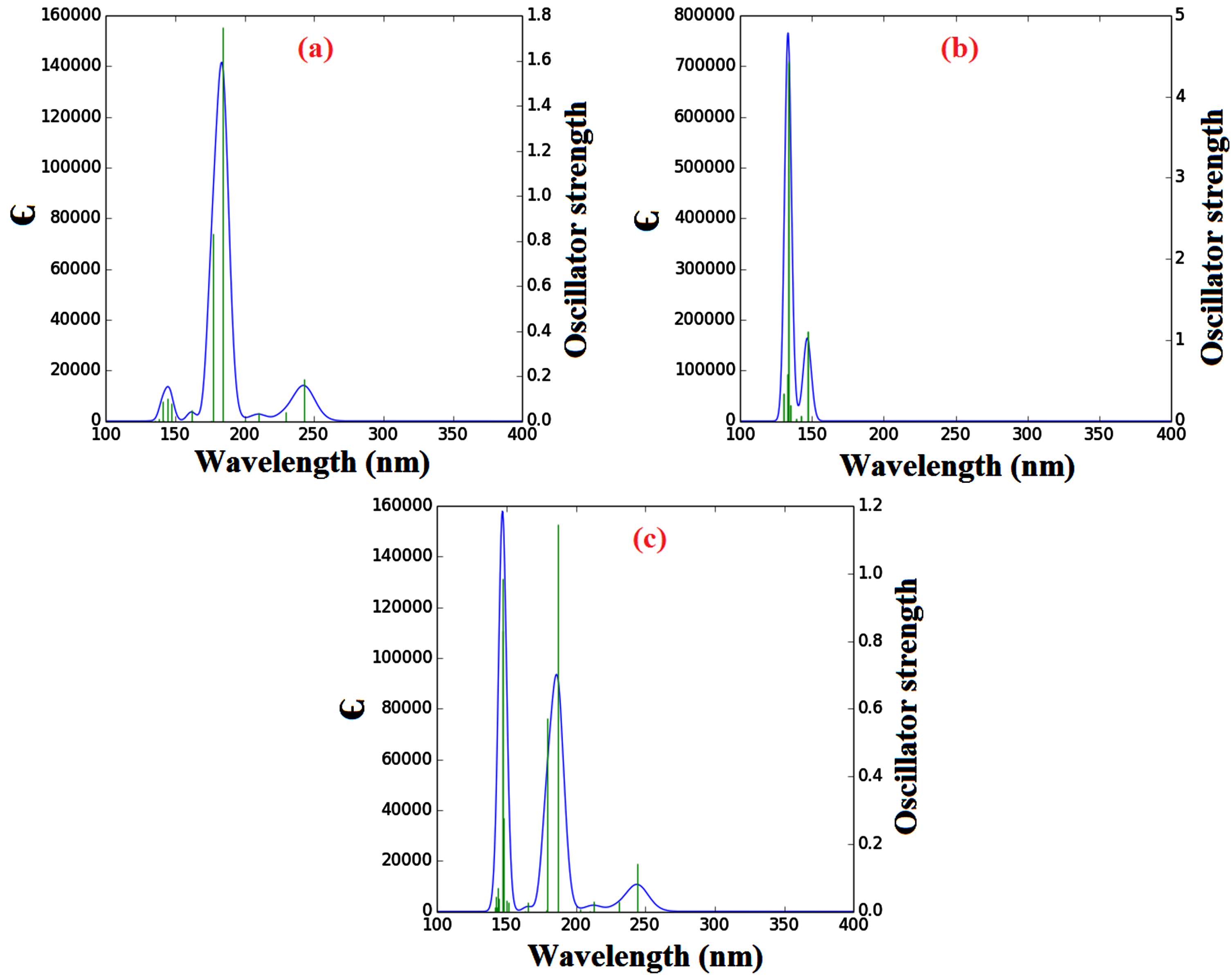
UV-Vis spectra of the: (a) DBT, (b) BNNT; and (c) DBT@BNNS complex.
The UV spectra of investigated compounds are presented in Fig. 5.
The adsorption behavior of the dibenzothiophene (DBT) molecule upon the boron nitride nanosheet (BNNS) was predicted by the theoretical calculations. The change of geometric parameters of the BNNS and DBT approves the interaction of DBT with the BNNS. The Eads and ΔHads values show that the adsorption of DBT over BNNS is an exothermic process. With the adsorption of DBT over the BNNS, the Eg value and conductivity are reduced. NBO analysis and charge difference (ΔN) show that the charge transfer in the DBT@BNNS complex occurs essentially from the DBT to the BNNS. After the interaction of DBT with nanosheet, the change of λmax of the DBT@BNNS complex confirms the interaction of DBT with BNNS. According to QTAIM analysis, topological parameters display an electrostatic interaction between BNNS and DBT, and the maximum amount of interaction energy is observed between the N40 and S5 atoms. The results of NICS analysis display that after the adsorption of DBT over the BNNS, all three rings A, B, and C of DBT have become more aromatic. We hope that our findings can be used for modeling and designing a suitable adsorbed for the adsorptive desulfurization process.
Footnotes
Funding
Not applicable.
Conflicts of interest/Competing interests
The authors declare no conflict of interest.
Availability of data and material
We understand that my manuscript and associated personal data will be shared with Research Square for the delivery of the author dashboard.
Code availability
Not applicable.