
Abstract
A novel, highly efficient, eco-friendly, and quick microwave-assisted synthesis of 2, 4, 5- substituted imidazoles by one-pot cyclo condensation of acenaphthoquinone, aromatic aldehydes, and ammonium acetate was reported under solvent-free conditions using ammonium chloride as a catalyst with excellent yield. The present methodology offers several advantages, such as mild reaction conditions, easy workup, short reaction time, excellent yield, purification of products by the non-chromatographic method, and reproducibility in large-scale synthesis. Compounds were characterized by IR, 1HNMR, 13C NMR spectroscopy and their melting points were compared with the literature reports.
Introduction
Microwave-assisted organic synthesis has revolutionized organic synthesis. Traditional thermal methods can create small molecules in a fraction of the time [1]. As a result, this technique has rapidly gained acceptance as a valuable tool for accelerating organic molecule synthesis and development processes [2, 3].
It is clear that the applications of microwave technology to the rapid synthesis of biologically significant heterocyclic molecules under solvent-free conditions are very promising and numerous. This technology has recently been recognized as a useful tool for a drug discovery program, especially in combinatorial chemistry. It is well known that the heterocyclic system is an important structural element in medicinal chemistry, showing a broad spectrum of pharmacological activities. We have selected several topics related to heterocyclic chemistry. Many of them are linked to interesting pharmacological properties (antiviral, including anti-HIV, anti-parasitic, antihistaminic, anti-cancer, anti-malarial, ..., etc.) [4–6].
Recently, considerable attention has been drawn to new combinatorial methodologies to develop new methods for organic synthesis. In this sense, multicomponent reaction (MCR) is the process in which three or more reactive substrates are combined together either sequentially or simultaneously in one vessel or in a single operation to give a target molecule scaffold without the isolation of any intermediate or changing the reaction conditions [7–10]. MCRs are a grateful class of reactions that apply to biological and pharmacological active compounds and successfully add to the large number of tools available to the modern chemist [11].
An important focus of current synthetic research is improving their rapidity, facility, chemical yield, selectivity, purity of product, and safety, as well as the tremendous scope of automation with advent of new microwave-assisted synthesis technology (MAOS). This methodology, performed in the absence of solvent, provides an environmentally clean technique that allows for high product yields at reduced energy costs [12, 13]. Microwave activation, as an unconventional energy source, can be used as an alternative to classical methods that enable the development of scale-up operations that are essential for the drug discovery process [1–6].
The development of new MCRs and the improvement of known MCRs, especially with microwave-assisted synthesis, are therefore of considerable current interest. One such reaction is the synthesis of substituted imidazoles via the synthesis of imidazole core, starting from the 1,2-dicarbonyl compound, aldehyde, and ammonia to obtain 2,4, 5-triphenylimidazole [14–19].
Heterocyclic compounds with the imidazole moiety have a wide range of biological activities, such as glucagon receptor [20], inhibitor of P38 MAP Kinase [21], canabinoid receptor antagonist [22], modulator of p-glycoprotein (p-gp) mediated multidrug resistance (MDR) [23], plant growth regulator [24], and a wide variety of therapeutic applications, such as anti-tumor [25], anti-bacterial [26], anti-inflammatory [27], fungicides [28], and anti-allergic agents [29]. Several modified methods have been reported for the synthesis of imidazole derivatives with various catalysts, such as trifluoroacetic acid (TFA), [30] [C4(mim)2](FeCl4)2 magnetic dicationic ionic liquid, [31] poly (4-vinylpyridinium Butane Sulfonic Acid) [32], HNO3@ nano SiO2, [33] L-Cysteine [34], and K5COW12O40.3H2O [35]. Each of the method mentioned for this reaction has its own advantages, while some of these methods have limitations, such as low yields, tedious workups, and effluent pollution. Therefore, the development of new mild and green methods to overcome these limitations still remains a challenge for researchers.
Clean technology innovates tight legislation on the emission of hazardous pollutants, including the use of alternative heterogeneous catalysts in chemical processes. In this regard, ammonium chloride (NH4+Cl–) is a very inexpensive, eco-friendly, available, and efficient catalyst and has been reported as a catalyst for the synthesis of various heterocyclic compounds.
In continuation of our works in the development of new methods in the synthesis of heterocyclic compounds, [17–19] we wish to report the multicomponent condensation of acenaphtoquinone, aromatic aldehydes, and ammonium acetate in the presence of ammonium chloride as a catalyst under solvent-free conditions in microwave irradiation.
Experimental
Material and method
All chemicals were purchased from Aldrich and Merck Chemical Company and were used without further purification. Melting points were determined on an electrothermal type 9100 melting point apparatus and are uncorrected. The IR spectra were recorded on a thermo Nicolet AVATAR-370 FT-IR spectrophotometer, and 1HNMR spectra were obtained on a Bruker DRX 300 spectrometer. Experiments were performed in a closed-vessel multi-mode Microsynth Milstone laboratory microwave oven using a 900-watt Westpointe microwave operating at 3.67 GHz with an internal volume of 0.9 m. All the products are known compounds, which were characterized by IR and 1HNMR spectra data and their melting points compared with the literature report.
General procedure for the synthesis of 2,4, 5- trisubstituted imidazole
In a beaker, a mixture of acenaphtoquinone (1 mmol), aromatic aldehyde (1 mmol), ammonium acetate (5 mmol), and ammonium chloride (3 mmol) as catalyst was thoroughly mixed and exposed to microwave irradiation (150 w, 20% power) for 4 minutes. Then the reaction mixture was monitored by tlc (EtOAc/Hexane: 2/1). After completion, the reaction mixture was cooled to room temperature and then washed with water (for the removal of ammonium chloride), and a crude product was precipitated. For further purification, it was re-crystallized with 96% ethanol to afford pure products.
Characteristic data for synthesized compounds
Phenyl-7H-acenaphtho[1,2-d]imidazole (Tables 2, 3a)
Yield: 90% ; m.p = 270°C; IR (KBr): 3200 (C-H), 1540 (C = N), 1340 (C = C). 1H NMR (300 MHz, DMSO-d6): 7.63–7.5(m, 7H), 7.7–7.9 (m, 5H) 12.4, (S, 1H). 13C NMR (100 MHz, DMSO-d6): 133.1, 132.5, 129.3, 127.8, 127.2, 126.5, 125.5, 124.1, 124, 123.4, 122.2, 120.8, 119.8.
8-(4- nitrophenyl)-7H-acenaphto [1,2-d] imidazole (Tables 2, 3b)
Yield: 96% ; m.p = 240°C; IR (KBr): 3180 (N-H), 1545 (C = N), 1340(C = C), 1383 (NO2) Cm–1. 1H NMR (300 MHz, DMSO-d6): 7.7–7.3(m, 7H), 8.1 (d, 1H), 8.6 (d, 1H), 8.3 (5, 1H), 12.2 (S, 1H, N-H). 13C NMR (100 MHz, DMSO-d6): 148.2, 148.0, 131.6, 131.3, 129.5, 128.7, 128.1, 127.3, 126.6, 123.4, 121.4, 120.3.
8-(3- nitrophenyl)-7H-acenaphto [1,2-d] imidazole (Tables 2, 3c)
Yield: 97% ; m.p = 229°C; IR (KBr): 3195 (N-H), 1535 (C = N), 1345(C = C), 1383 (NO2) Cm–1. 1H NMR (300 MHz, DMSO-d6): 7.63–7.5(m, 7H), 8.2 (d, 1H), 8.4 (d, 1H), 8.8 (5, 1H), 12.69 (S, 1H, N-H). 13C NMR (100 MHz, DMSO-d6): 148.8, 148.0, 132.8, 131.1, 129.6, 128.9, 128.2, 127.5, 127.1, 122.8, 120.4, 119.3.
8-(4- cyanophenyl)-7H-acenaphto [1, 2-d] imidazole (Tables 2, 3d)
Yield: 98% ; m.p = 203°C, IR (KBr): 3400 (cyanide), 3200 (N-H), 1543 (C = N), 1344(C = C) Cm–1. 1H NMR (300 MHz, DMSO-d6): 7.8–7.4(m, 7H), 8.1 (d, 1H), 8.5 (d, 1H), 8.1(5, 1H), 12.4 (S, 1H). 13C NMR (100 MHz, DMSO-d6): 148.9, 148.2, 131.9, 131.1, 129.2, 128.4, 128.1, 127.3, 126.6, 123.4, 121.4, 120.3.
8-(2- bromo-phenyl)-7H-acenaphto imidazole [1,2-d] (Tables 2, 3e)
Yield: 93% ; m.p = 296°C; IR (KBr): 3100 (N-H), 1650 (C = N), 1281 (C = C) cm–1, 1040 (C = Br) (1H NMR (300 MHz, DMSO-d6): 7.5 (d, 1H), 7.1 (d, 1H), 7.4–7.6 (m, 1H), 7.7–7.8 (t, 3H), 7.9 (d, 1H), 12.2 (S, 1H). 13C NMR (100 MHz, DMSO-d6): 132.9, 131.5, 130.6, 130.2, 128.8, 128.1, 127.7, 127.2, 126.8, 123.5, 121.4, 120.2.
8-(3- bromo-phenyl)-7H-acenaphto imidazole [1,2-d] (Tables 2, 3f)
Yield: 94% ; m.p = 289°C; IR (KBr): 3110 (N-H), 1638 (C = N), 1276 (C = C) Cm–1, 1030 (C = Br) (. 1H NMR (300 MHz, DMSO-d6): 7.8 (d, 1H), 7.2 (d, 1H), 7.7–7.5 (m, 2H), 7.9 (d, 1H), 12.2 (S, 1H). 13C NMR (100 MHz, DMSO-d6): 132.9, 131.5, 130.6, 130.2, 128.8, 128.1, 127.7, 127.2, 126.8, 123.5, 121.4, 120.2.
8-(2- chloro-phenyl)-7H-acenaphto imidazole [1,2-d] (Tables 2, 3g)
Yield: 98% ; m.p = 190–191°C; IR (KBr): 3039.8 (N-H), 1694 (C = N), 1281 (C = C) cm–1. 1H NMR (300 MHz, DMSO-d6): 7 (d, 1H, Ar), 7.2 (d, 1H), 7.4–7.6 (m, 1H), 7.7–7.8 (t, 3H), 7.95 –7.98(d, 1H), 12.69(S, 1H).13C NMR (100 MHz, DMSO-d6): 132.7, 131.6, 130.8, 130.4, 129.5, 128.7, 128.1, 127.9, 126.9, 121.7, 120.4, 119.3.
8-(3- methoxy-phenyl)-7H-acenaphto imidazole [1,2-d] (Tables 2, 3h)
Yield: 83% ; m.p = 269°C IR (KBr): 3100 (N-H), 2965 (C-H), 1630 (C = N), 1265 (C = C) cm–1 (1H NMR (300 MHz, DMSO-d6): δH = 7.6 (d, 1H), 7.2 (d, 1H), 7.7–7.5 (m, 2H), 7.9 (d, 1H), 4.5 (S, 3H), 12.1 (S, 1H). 13C NMR (100 MHz, DMSO-d6); δ= 133.3, 131.2, 130.6, 130.2, 128.2, 128.1, 127.3, 127.2, 126.8, 123.5, 121.4, 120.2, 75.
8-(4- methoxy-phenyl)-7H-acenaphto imidazole [1,2-d] (Tables 2, 3i)
Yield: 85% ; m.p = 249°C IR (KBr): 3130 (N-H), 2950 (C-H), 1625 (C = N), 1250 (C = C) cm–1. 1H NMR (300 MHz, DMSO-d6): δH = 7.7 (d, 2H), 7.3 (d, 2H), 7.7–7.5 (m, 2H), 7.9 (d, 1H), 4.5 (s, 3H) 12.3 (S, 1H). 13C NMR (100 MHz, DMSO-d6); δ= 133.5, 132.3, 131.8, 130.1, 128.7, 128.1, 127.6, 127.1, 126.5, 124.5, 122.5, 120.1, 77.
8-(3- methyl- phenyl)-7H-acenaphto[1,2-d] imidazole (Tables 2, 3j)
Yield: 87% ; m.p = 230°C; IR (KBr): 3420 (N-H), 2910 (C-H), 1620 (C = N), 1259 Cm–1. 1H NMR (300 MHz, DMSO-d6): 7.6–7.5(m, 7H), 7.8 (d, 1H), 7.9 (d, 1H), 8 (m, 3H), 12.69(S, 1H). 13C NMR (100 MHz, DMSO-d6): 139.2, 136.7, 133.3, 131.1, 129.2, 128.3, 128.2, 126.8, 126.5, 125.8, 122.3, 120.1, 119.7, 70.
8-(4- methyl- phenyl)-7H-acenaphto[1,2-d] imidazole (Tables 2, 3k)
Yield: 88% ; m.p = 231–233°C; IR (KBr): 3420 (N-H), 1606 (C = N), 1259 Cm–1. 1H NMR (300 MHz, DMSO-d6): 7.63–7.5(m, 7H), 8.2 (d, 1H), 8.4 (d, 1H), 8.8 (5, 1H), 12.69(S, 1H). 13C NMR (100 MHz, DMSO-d6): 138.1, 136.3, 131.5, 131.2, 129.2, 128.2, 128.2, 126.8, 126.5, 125.2, 122.5, 120.1, 119.7, 74.
Results and discussion
In continuing our interest in microwave-assisted synthesis of novel imidazole derivatives, we developed a one-pot reaction catalyzed by ammonium chloride. In the present work, we find that ammonium chloride is a highly efficient catalyst for the one-pot synthesis of substituted imidazoles from acenaphtenequinone, aromatic aldehydes, and ammonium acetate under microwave-assisted, solvent-free conditions (scheme 1). The multicomponent nature of the procedure and the existence of a wide variety of commercially available aromatic aldehydes, acenaphtoquinone, and ammonium acetate/ammonium chloride make this reaction an ideal candidate for the combinatorial synthesis technology.
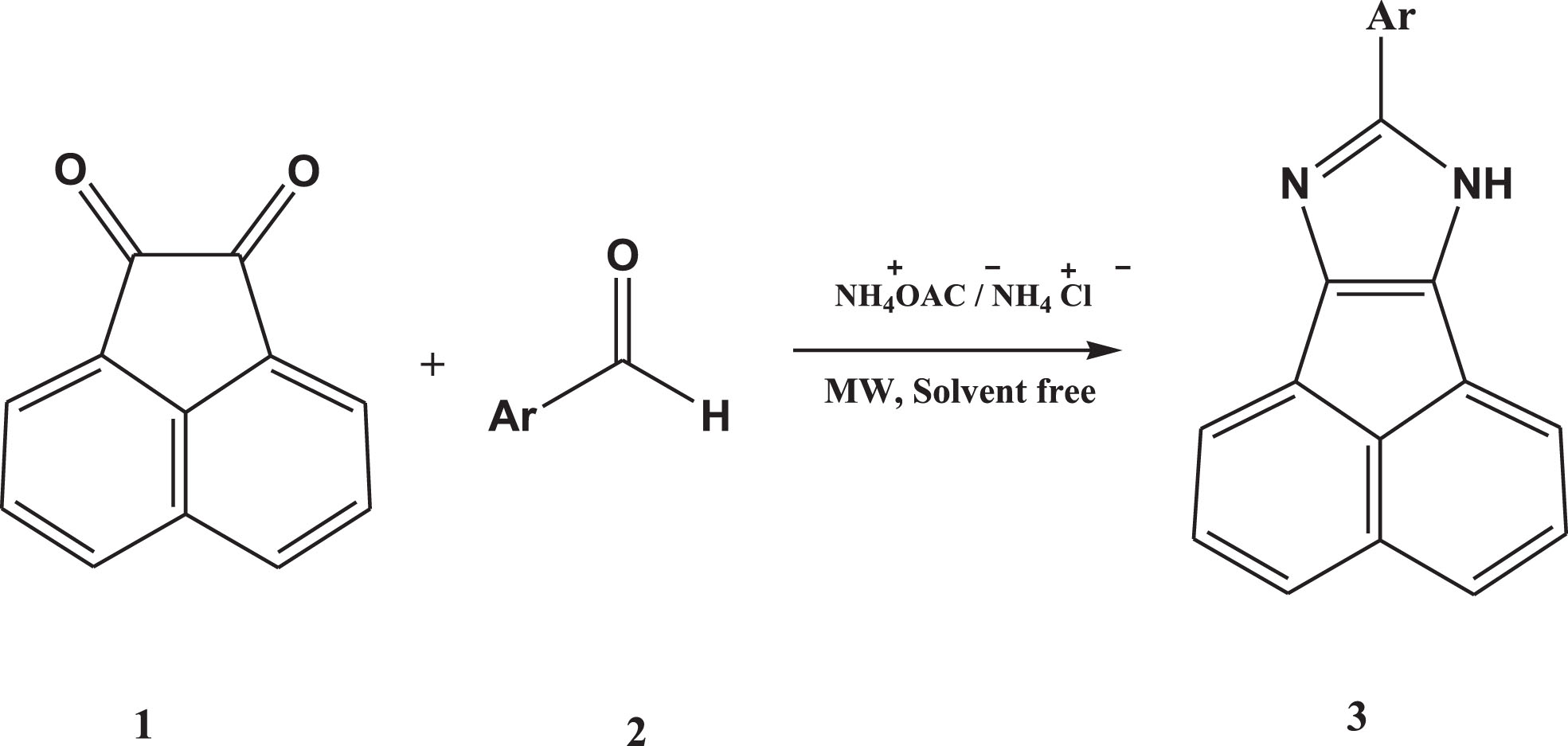
One-pot three component synthesis of substituted imidazoles in the presence of ammonium acetate/ ammonium chloride as a catalyst in MW irradiation and solvent-free condition.
To find the best condition for this reaction (Scheme 1), we examined the efficiency of different reaction media and catalyst amounts for the condensation reaction. Acenaphthoquinone, benzaldehyde, and ammonium acetate/ammonium chloride were chosen as model reactions (Table 1). For each reaction condition, the conversion of initial reactants to acenaphto[1,2-d] imidazole was followed by tlc. Reactions at different conditions and various molar ratios of substrates in the presence of ammonium chloride indicate that the best condition was a molar ratio of acenaphthoquinone/benzaldehyde/ ammonium acetate/ammonium chloride of 1:1:5:3 under solvent-free conditions and microwave irradiation.
Effect of catalyst on reaction time and yield

All reactions were run using acenaphthoquinone/aromatic aldehyde/ammonium acetate/ammonium chloride molar ratio 1:1:5:3.
Synthesis of Imidazoles 3a-k under Solvent free Condition and Microwave Irradiation

The optimal conditions were then applied for the preparation of a series of substituted imidazoles 3a-k. The product yields are shown in Table 2. It is interesting to mention that this method is useful for preparation of substituted imidazoles using different aromatic aldehydes with a wide range of ortho, meta and para substituted aldehydes without side product formation. This method not only affords the product an excellent yield, but also avoids the limitations associated with catalysts like cost, handling, safety, and pollution. It is to be mentioned that the obtained products could be easily separated because ammonium chloride is completely soluble in water at room temperature, but the obtained products are not, so the obtained products were separated by simple filtration. All known products 3a-k were characterized by their 1HNMR, IR, 13CNMR and their physical properties were compared with previously reported data. Our obtained results and conditions for the synthesis of acenaphto [1,2-d] imidazole (3a) were compared with previously reported data in Table 3. The results show that our method is significantly comparable with the former methods in yield and reaction time (Table 3).
Synthesis of acenaphto[1,2-d] Imidazoles 3a using different catalyst and reaction condition

(4- SB)T(S-Ph)PHSO4; 4- (Sulfobutyl) tris (4-sulfophenyl) phosphonium hydrogen sulfate. P- TsOH; P-toluenesulfonic acid. P(4VPBSA) HSO4: Poly (4-vinylpyridinium Butane Sulfonic Acid). K5COW12O40.3H2O; Potassium Dodecatugstocobaltate trihydrate.
The proposed mechanism for the synthesis of imidazole derivatives (5) (Scheme 2) can be the initial simultaneous addition of two amino groups to a protonated carbonyl group (1) (that is protonated by ammonoium chloride) for the formation of di amino intermediate (2). Then, this intermediate (2) reacted with protonated carbonyl groups of acenaphtenequinone (3) along with the elimination of two H2O for the formation of diimidazole derivatives (4) which resulted in the production of the final imidazole derivatives (5) by [1,5]-H-shift.

Proposed Mechanism for acenaphto [1,2-d]Imidazole Derivatives synthesis.
We have developed a remarkably accelerated and environmentally friendly procedure for the synthesis of novel substituted imidazoles via one-pot three-component condensation of acenaphthoquinone, aromatic aldehydes, and ammonium acetate/ammonium chloride under microwave-assisted, solvent-free conditions. The major advantages of this methodology compared to previous methods are short reaction times, easy workup, clean reaction profile, excellent yield, and the purification of compounds by the non-chromatography (re-crystallization only) method. Therefore, it is believed that this approach could be of great value to other novel synthetic processes in the field of imidazole derivatives.
Footnotes
Acknowledgments
The author wishes to thank the Ahvaz Branch, Islamic Azad University for the financial support.