
Abstract
Renal cystic diseases are a clinically and genetically diverse group of renal diseases that can manifest in utero, infancy, or throughout childhood and adulthood. These diseases may be unilateral or bilateral with a single cyst or multiple cysts, or with increased echogenicity of the renal cortex without macroscopic cysts. Certain cystic renal diseases are life-threatening, with many developing chronic kidney and hepatic disease if not recognized early enough. Therefore, due to the prevalence and life-altering complications of this specific group of diseases in vulnerable populations, it is crucial for clinicians and healthcare providers to have an overall understanding of cystic diseases and how to pre-emptively detect and manage these conditions. In this review, we discuss in detail the epidemiology, genetics and pathophysiology, diagnosis, presentation, and management of numerous genetic and sporadic renal cystic diseases, such as polycystic kidney disease, multicystic dysplastic kidney, and calyceal diverticula, with an emphasis on prenatal care and pregnancy counseling.
Introduction
Polycystic kidney diseases (PKDs) are a clinically and genetically diverse group of renal diseases that can manifest in utero, infancy, or throughout childhood and adulthood. PKDs may be unilateral or bilateral with a single cyst or multiple cysts, or with increased echogenicity of the renal cortex without macroscopic cysts. PKDs specific to the neonatal period are clinically significant as they contribute to the development in neonates and infants. PKDs may be either genetic or sporadically occur. Genetic cystic kidney diseases represent a group of slowly progressive, chronically debilitating disorders that are one of the most common causes of end-stage renal disease in childhood [1]. This group of genetic cystic kidney diseases is mainly comprised of autosomal recessive polycystic kidney disease (ARPKD), autosomal dominant polycystic kidney disease (ADPKD), glomerular cystic kidney disease, Bardet-Biedl syndrome (BBS), nephronophthisis-medullary cystic kidney disease complex, and hepatocyte nuclear factor-1beta nephropathy [2]. Sporadic renal cystic diseases consist of multicystic dysplastic kidney (MCDK), calyceal diverticula (CD), simple renal cysts, and complex renal cysts [3]. CD has also been associated with certain genetic mutations. While CD and MCDK are generally not inherited renal cystic diseases, and are most often sporadic in nature, they have been linked to certain genetic disorders. Specifically, renal dysplasia has been associated with genetic syndromes such as Meckel-Gruber syndrome, Vertebral defects-Anal atresia-Cardiac defects-Tracheo-esophageal fistula-Renal anomalies-Limb abnormalities (VACTERL), Renal-hepatic-pancreatic dysplasia (RHPD), and splenic disorders [4].
The incidence of cystic kidney diseases can vary from 0.44 cases per 10,000 births for genetic polycystic kidney disease (such as ARPKD and ADPK) to 4.1 cases per 10,000 births for sporadic renal cystic diseases such as multicystic dysplastic kidney (MCDK) [5]. Certain cystic renal diseases are life threatening, and some may go on to develop chronic kidney and hepatic disease. Due to the prevalence and life-altering complications of this specific group of diseases in vulnerable populations, it is imperative for clinicians and healthcare providers to pre-emptively detect and manage these conditions.
ADPKD is the most common form of inherited PKD. ADPKD may be diagnosed in any age and is associated with progressive cyst formation. Although it is a very rare cause of end-stage renal disease in children, it accounts for 5% of ESRD seen later in life for adults [6]. ARPKD presents as a hepatorenal fibrocystic disorder that can present prenatally, and can be fatal if severe pulmonary hypoplasia due to oligohydramnios is present [2]. In comparison, fsporadic renal cystic diseases may also present neonatally. MCDK is the most common form of cystic renal dysplasia with an incidence of 1:4300 live births, and can also be fatal if present bilaterally [4]. CD is a less common presentation of non-genetic renal cystic diseases, and may present at any age with renal diverticula that may lead to stones or abscesses [7].
Fifteen to twenty percent of all congenital abnormalities identified on ultrasound during pregnancy are due to malformations of the genitourinary tract [8]. These abnormalities may manifest as cystic, dysplastic, or obstructive changes to the kidney, ureter, or bladder. Renal cystic diseases may be identified on ultrasound prenatally as early as 17 weeks, presenting with hyperechogenic kidneys or pyelectasis [4]. Renal cystic kidney diseases may be due to an inherited gene defect or to a structural anomaly leading to an obstruction or malformation. This review will specifically focus on ARPKD, ADPKD, MCDK and CD with emphasis on prenatal care and pregnancy counseling.
Prenatal care and evaluation of renal cystic diseases
The fetal excretory system begins as the primitive pronephros in the third week of development and regresses as the metanephros form in week five. By the 6th week of development, the fetal kidneys begin as the mesonephric duct, which develops as the ureteric bud, near the cloaca, grows cephalad to form the ureter and collecting system of the renal pelvis. The metanephrogenic blastema then forms renal tubules and glomeruli and begins to make hypotonic urine by 16 weeks. Using ultrasound imaging, the fetal bladder can be visualized at 9 weeks and the fetal kidneys at 11-12 weeks of gestation, respectively. During the first trimester, the kidneys are hyperechoic oval structures on both sides of the spine. Their echogenicity decreases throughout gestation, while the corticomedullary differentiation begins at approximately 14 weeks. The kidneys should grow at a rate of 1.1 mm each week [9].
Fetal renal anomalies may involve renal number (absent, duplicated, or pelvic kidneys), size (small or enlarged kidneys or collecting systems), or appearance (hyperechoic or hypoechoic/cystic). While some anomalies, such as renal agenesis, are apparent at any point in gestation, others may evolve throughout the pregnancy. When evaluating the fetal urinary tract, the size, location, and echogenicity of the kidneys should be described. The kidneys should be viewed in the sagittal plane to evaluate growth and in the transverse plane to evaluate the renal pelvis, which should measure 4 mm or less in the second trimester and 7-8 mm or less in the third trimester. The ureters are normally not visualized. The bladder should be seen between the umbilical arteries and fills approximately every 30 minutes. When full, the bladder should measure 3 cm or less in the second trimester and 5 cm or less in the third trimester. The bladder wall thickness should be less than 3 mm when full. In addition, the amniotic fluid index should be measured as approximately 2/3 of the fluid is comprised of fetal urine [10].
Congenital anomalies of the kidneys and urinary tract (CAKUT) are one of the most common malformations seen on prenatal ultrasound. Dilatation of the renal pelvis, small or large hyperechogenic kidneys, and cystic kidneys represent some of the most common findings associated with CAKUT [8]. When a cystic disease is suspected, the size, number, appearance, and location of the cyst(s) should be described along with the appearance of the remaining renal tissue and contralateral kidney. The most common finding is pyelectasis. Pyelectasis is graded based on severity (0-normal, I-mild dilation of pelvis, II-moderate splitting of the pelvis and calyces, III-marked splitting, pelvis dilated outside the renal borders and calyces dilated, IV-further pelvicalyceal dilation with thinned renal parenchymal thickness). When there is an early obstruction to the urinary tract, the grade IV pyelectasis resembles multicystic dysplastic kidneys, where no normal renal parenchyma is noted with multiple large cysts (“bunch of grapes”). The differential diagnosis in these cases includes early uretero-pelvic junction obstruction, autosomal dominant polycystic kidney disease, Meckel-Gruber syndrome, autosomal recessive polycystic kidney disease, and trisomy 13. In cases of bladder outlet obstruction, amniocentesis can be helpful in determining residual renal function. Fetal MRI is may be indicated, especially in cases of echogenic kidneys.
Pregnancy counseling
When a renal anomaly is diagnosed on imaging, the patient should be offered a consultation with a maternal fetal medicine specialist and neonatologist, depending on the anomaly. When genetic testing is being considered, a referral to a genetic counselor should be obtained. The visit will review the ultrasound findings, the expected prognosis if known, the potential for recurrence, and the pregnancy management plan. The genetic counselor will review the family history, including a history of renal or hepatic disease, unexplained stillbirth, pregnancy loss, history of need for dialysis, hypertension, or other congenital disorders and inquire about consanguinity. Options for prenatal and postnatal genetic testing can be reviewed as well as assistance with interpretation of any abnormalities identified on genetic testing. The genetic counselor can also assist in identifying at risk relatives and coordination of testing and renal ultrasounds of family members if applicable. The specific testing offered to the patients will depend on the renal findings as well as the presence of other anomalies. For example, testing for TORCH titers and ARPKD in a fetus with echogenic kidneys or Beckwith-Wiedemann syndrome is a fetus measuring large for gestational age. If a mutation is known in the family, site specific testing is offered. As the genetics of renal disorders evolves, the clinical utility is likely to improve and can provide valuable information regarding clinical management, prognosis and future recurrence risks. Any pregnancy affected with renal disease should be offered genetic counseling.
For conditions that affect bilateral kidneys and lead to oligo-anhydramnios, the fetus is at risk for Potter’s Sequence or severe fetal lung hypoplasia, which is often hardly compatible with extra-uterine life. In these cases, the option of pregnancy termination should be offered. If the pregnancy is continued, then palliative care or hospice services should be offered and arranged if the fetus is deemed nonviable. In cases of suspected ARPKD, risk factors for early dialysis dependency should be discussed with parents as well. In cases regarding CKD, such as hypertension or dialysis, a Pediatric Nephrologist is essential for providing guidance regarding prognosis to parents.
In less severe cases, the pregnancy should be monitored with serial ultrasounds to evaluate the fetal kidneys, ureters, and bladder, the amniotic fluid index, and the fetal growth every 4–8 weeks. Based on the prenatal imaging, the parents should be prepared for a range of possibilities from a fetus that may do well and be discharged home to one that may need comprehensive care in the neonatal period, up to and including neonatal dialysis. In some cases, the neonates may survive only a few weeks. In cases of autosomal dominant or recessive polycystic kidney disease, the parents should be offered testing in order to assess their recurrence risk, health, and for possible extra-renal abnormalities such as liver cysts.
Sporadic renal cystic diseases
Multicystic dysplastic kidney (MCDK)
Epidemiology
MCDK is the most common form of cystic renal dysplasia in children and part of the wide spectrum of early developmental disorders often summarized as CAKUT (congenital anomalies of the kidney and urinary tract). MCDK is a congenital malformation of the kidney identified by multiple cysts without significant identifiable renal parenchyma [4]. In MCDK, the kidneys are abnormally developed and have poorly differentiated nephrons, increased stromal components with associated cystic and sometimes metaplastic kidneys [8]. The condition occurs more commonly in males than females, but females are twice as likely to have bilateral MCDK. The left kidney is more often affected than the right kidney [11]. Unilateral dysplastic kidneys occur in approximately 1 in 4,000 fetuses, and bilaterally in 1 in 10,000 fetuses [8].
Genetics and pathophysiology
Multicystic kidney disease derives from a blockage in the urinary tract at some point between the renal pelvis and the urethra. This complete obliteration of urinary flow at a critical stage of development ultimately results in a non-functioning kidney [12]. The renal parenchyma is replaced by numerous smooth-walled cysts that do not communicate with the renal pelvis. The renal parenchyma is also surrounded by echogenic cortex with an atretic ureter. There are two subtypes of MCDK that are classified based on their gross appearance. One form is solid cystic dysplasia and the other form is hydronephrotic. Solid cystic dysplasia has increased stromal components and smaller cysts, while the hydronephrotic form has an identifiable renal pelvis. Usually unilateral MCDK is nonhereditary and sporadic, but there have been rare cases attributed to an autosomal dominant inheritance pattern [4]. While unilateral kidney dysplasia is commonly sporadic, bilateral dysplasia may indicate the presence of aneuploidy or inherited genetic conditions with potential mutations in a number of different genes involved in early development. If bilateral kidney disease is detected prenatally, then this may be associated with low amniotic fluid, potentially leading to Potter’s sequence [8].
Presentation and diagnosis
Dysplasia in the antenatal period will present as large bright kidneys with or without the presence of visualized cysts, and the degree of abnormality in the echogenic kidneys will be the key prognostic indicator for MCDK. If cysts are present at 20 weeks or earlier, it is more likely to be MCDK as cysts originating from other cystic kidney diseases usually only show up on ultrasound later in pregnancy [8]. Neonates may present with a small kidney with few or no cysts in the corticomedullary region. They may also present with polyuria, polydipsia, salt-wasting, juvenile gout, and will ultimately go on to develop ESRD in adulthood [3].
Patients presenting with cystic renal dysplasia secondary to nephronophthisis-related ciliopathies (NPHP-RC) may present early during childhood or adolescence. Patients with NPHP-RC may present with cystic-fibrotic kidney disease, brain development defects, retinal degeneration, skeletal deformities, obesity, facial dimorphism, and congenital heart disease [13].
Bilateral MCDK may have poor prognosis for survival. MCDK has been known to occur with various genetic malformations and extrarenal features. The list of organs potentially involved in these cases is long and include differential diagnoses such as Bardet-Biedl syndrome, Zellweger syndrome, VACTERL, renal coloboma syndrome, prune belly syndrome, branchio-oto-renal syndrome, and renal-hepatic-pancreatic dysplasia [4]. As discussed, there may be associated genital tract anomalies particularly in females such as HNF1β-related, or Mayer-Rokitansky’s disease [3].
MCDK is now detected earlier with increased use of antenatal ultrasounds, as previously it would commonly present as an incidental finding with a palpable abdominal mass. Viable MCDK may present unilaterally over long periods of time, and will cause the unaffected kidney to become hypertrophied as it compensates for the loss of the other kidney (Fig. 1). This compensation may result in vesicoureteral reflux or ureteropelvic junction obstruction [14].

Ultrasound imaging of multicystic dysplastic kidney (MCDK). A. Sagittal right kidney ultrasound scan of fetal abdomen shows multiple round cysts, non-communicating with absence of renal parenchyma. Identifiable renal sinus with irregularly contoured kidney. B. Transverse view of an enlarged fetal kidney with an irregular surface and loss of differentiation in the renal parenchyma consistent with a multicystic dysplastic kidney.
Ultrasound is the most efficient imaging modality for diagnosis. Both prenatal and postnatal renal ultrasounds, as well as diuretic renograms are important to diagnose and determine the extent of renal tissue damage [3]. Some institutions are using MRI more frequently to evaluate abnormal kidneys when ultrasound is technically challenging due to maternal habitus or fetal position 8. Current consensus regarding imaging is for ultrasound to the main method for assessing pediatric renal cysts, with selected indications for MRI and contrast-enhanced US [15]. Limited radiographic evidence is preferable. If concern for contralateral anomalies, further workup with either VCUG or nuclear medicine studies should be directed by any abnormalities see on renal ultrasound or after developing clinical sequelae, from obstructive uropathy or VUR in contralateral kidney, such as hypertension, UTI, or palpable abdominal masse [14].
If detected antenatally, bilateral dysplasia will likely have a very poor outcome, especially if there is significant oligohydramnios and decreased kidney growth during pregnancy. With age, patients may receive drug therapy, renal replacement therapy, or require a renal transplant when the patient achieves appropriate weight [8]. The majority of cases either involute partially or completely, causing some to disregard follow-up and monitoring. While there is an extremely rare chance of developing a renal malignancy in these patients, prophylactic nephrectomy is not indicated, and are monitored by screening ultrasound instead. Both the affected and functional kidneys’ sizes should be monitored closely with serial ultrasounds. Patients should also be monitored for the presence of a vesicoureteral reflux (VUR), as this can lead to urinary tract infections which can threaten outcomes in the functionally contralateral kidney [4, 12]. Routine blood pressure monitoring assessing for HTN should be done for children with MCDK and appropriately managed, if present. If no other etiologies are identified for the hypertension, it is possible that nephrectomy may cure the HTN [16].
Urological and nephrological follow up is only warranted for the contralateral kidney in those with “complex” MCDK [16]. Associated anomalies in the contralateral kidney have been found to occur in 33% of MCDK patients, with VUR occurring in 20% of patients, and ureteropelvic junction and ureterovesical obstructions occurring less commonly [14].
Calyceal diverticulum
Epidemiology
Calyceal diverticulum (CD) is a rare event in the pediatric population and presents as an outpouching of the transitional epithelial lined collecting system. These outpouchings, or diverticula, may result in stone formation and infection. CD may be found at any age, and may present on either the left or right side [17]. Renal tract malformations account for 40% of children with end-stage renal disease [18]. CD are found in 0.21-0.6% of intravenous urograms (IVU) performed on adults, with a similar prevalence in children, and appear to affect women more than men [7].
Genetics and pathophysiology
The exact etiology of CD development is unknown, but is thought to be due to either congenital ureteric bud regression failure as a result of inflammation or from a possible obstructive process in response to infection or reflux [19]. Studies have suggested CD occur early in development at approximately 35 days, or possibly just before birth. Several different potential causes of CD include obstructive, neuromuscular, traumatic or fibrotic causes [7]. While CD has not been strictly categorized as a genetic renal cystic disease, it has been associated with mutations in genes such as EYA1 or SIX1 causing a branchio-oto-renal syndrome [18].
Presentation and diagnosis
CD are classified as type I or type II. Type I CD communicate with a minor calyx or an infundibulum, while type II originate from the renal pelvis or major calyx, are larger in size, usually symptomatic, and located in the central part of the kidney [7]. While most CD are small or asymptomatic, patients may also present with flank pain, gross hematuria, or recurrent UTIs. CD initially appears as a simple or complex cyst on early initial ultrasound, and thus may be under diagnosed. Diagnosis is most optimal by visualizing the renal cystic lesion with retrograde contrast opacification from the collecting system by either intravenous urography or computed tomography urography (CTU) [7]. Pareek et al. performed a multi-phase contrast-enhanced CT of a benign calyceal diverticula (Fig. 5) [20]. One study showed that Tc-99m DTPA diuretic renal scan seems to be more sensitive than CTU in diagnosing CD [17].
Management and prognosis
There is no consensus within the pediatric literature regarding the optimal management for patients with calyceal diverticula. SWL is considered the first-line management when patients have stone-bearing diverticula (Table 1) [7]. One study was able to show that they can use the endoscopic approach as a first-line, minimally invasive option for patients with small, endophytic diverticula. Patients with large diverticula that are exophytic with thin overlying parenchyma, anteriorly located diverticula, or diverticula refractory to other forms of treatment should be considered for laparoscopic approach [7, 19].
Perinatal and neonatal renal cystic diseases
Perinatal and neonatal renal cystic diseases
MCDK, multicystic dysplastic kidney; CD, calyceal diverticulum; ARPKD, autosomal recessive polycystic kidney disease; ADPKD, autosomal dominant polycystic kidney disease; ESRD, end-stage renal disease; CAKUT, congenital anomalies of kidney and urinary tract; VACTERL, vertebral defects-anal atresia-cardiac defects-tracheo-esophageal fistula-renal anomalies-limb abnormalities; IVU, intravenous urograms.
Both ARPKD and ADPKD are inherited disorders that involve the presence of multiple cysts on both kidneys without the presence of dysplasia [3]. Both differ in their inheritance pattern, clinical presentations, and management [21]. Figure 2 shows a diagnostic algorithm for hereditary renal cystic diseases.
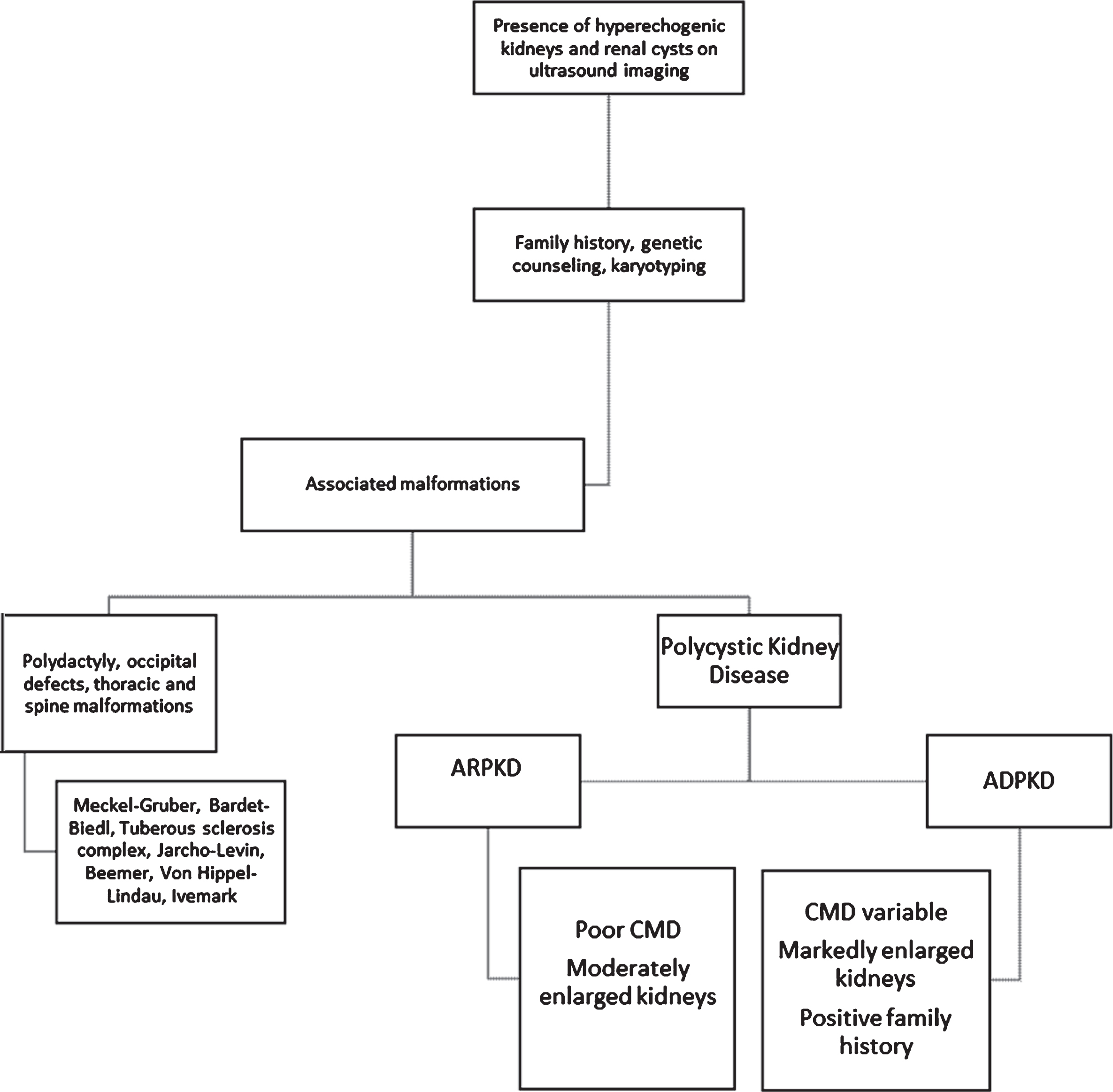
Diagnostic algorithm for hereditary renal cystic diseases. ARPKD, autosomal recessive polycystic kidney disease; ADPKD, autosomal dominant polycystic kidney disease; CMD, corticomedullary differentiation.
Epidemiology
ARPKD is a dual-organ disease involving both liver and kidney with an incidence of 1:20,000 to 1:40,000 (Table 1). ARPKD is a major cause of morbidity and mortality in neonates, infants, and young adults [1]. ARPKD is a rare, hepatorenal fibrocystic disease, characterized by non-obstructive, fusiform, cystic distension of renal collecting ducts and invariably, but varying degrees of congenital hepatic fibrosis (CHF) due to effects on epithelial cells lining the bile ducts known as ductal plate malformation (DPM). The initial symptoms and age at which these symptoms present may vary. Parents of these patients are heterozygotes (carriers), and are usually clinically unaffected by this disease due to its autosomal recessive inheritance pattern. Heterozygous carriers are known to be at increased risk for polycystic liver disease and renal involvement associated with medullary hyperechogenecity on ultrasound. It still needs to be determined if some of these individuals become symptomatic with age [22, 23]. All genders, ethnicities, and racial groups are equally affected in ARPKD [1]. Patients generally present with large echogenic kidneys during the neonatal period, but can present later in life as well. Infants that survive the neonatal period have a 50% chance of developing end-stage renal disease (ESRD) by age 10 years. The neonatal mortality from ARPKD nowadays seems to be lower than the 30–50% still found in the literature [3].
Genetics and pathophysiology
ARPKD is caused primarily by mutations in PKHD1, which encodes fibrocystin/polyductin (FPC), a protein associated with primary cilia on epithelial cells in the bile ducts and kidneys. When there is dysfunction in these cilia, there is cystic dilation of the renal collecting ducts with possible presentation of biliary dysgenesis. Hepatic abnormalities, such as periportal fibrosis, result from ductal plate malformations with cystic dilation of the intrahepatic and extrahepatic bile ducts. Truncating PKHD1 mutations on both alleles typically display a severe phenotype associated with a high rate of perinatal or neonatal death [1].
Due to a variety of possible pathogenic causes, the epithelial cells in PKD have different functions in comparison to normal renal epithelial cells; these epithelial cells have proliferative, secretory effects as opposed to their normal absorptive and non-proliferative features which allow for cystic growth in the kidneys [2, 4]. Histologically, every ARPKD patient demonstrates DPM with CHF, but only a minority of neonates are symptomatic. In addition, clinical features due to portal hypertension usually only occur during childhood and adolescence. Kidneys may have multiple small cysts confined to the collecting ducts that may extend into the renal cortex. Microscopic cysts can measure less than 2 mm in diameter, while the renal calyces, pelvis, and vessels appear normal in structure [1].
Presentation and diagnosis
Up to one half of diagnoses are made prenatally. At birth, patients may present with pulmonary hypoplasia as a result of oligohydramnios due to large cystic kidneys. During development, the lungs need adequate amniotic fluid to expand and develop. Inadequate amounts of amniotic fluid lead to diaphragmatic restriction and pulmonary hypoplasia (Potter’s sequence). Pulmonary insufficiency is the major cause of morbidity and mortality in neonates with ARPKD [1].
At birth, patients will present with large palpable flank masses, which can complicate delivery. Over time, these flank masses may decrease or remain stable in size [1].
Hyponatremia may be present initially, but will resolve if no indication of acute kidney injury is present. Patients may present with urinary concentrating defects such as polyuria and polydipsia [1].
Complications of congenital hepatic fibrosis (CHF) may develop at birth, or much more often they remain asymptomatic until childhood. CHF, and CHF complications such as biliary ductal ectasia (Caroli disease) must be closely monitored because the hepatobiliary disease may lead to portal hypertension. Portal hypertension is the main complication of CHF and presents with clinical signs of hepatosplenomegaly and hypersplenism with thrombocytopenia. Hypersplenism may result in variceal and hemorrhoid bleeding, and increased risk of developing bacterial infections from ascending cholangitis [1].
A small subset of older patients may present with more symptoms related to hepatic disease than renal disease. 40% of ARPKD survivors will present with severe renal and hepatobiliary disease. 60% of ARPKD survivors will present with either severe kidney and mild hepatobiliary disease, mild kidney and severe hepatobiliary disease, or mild kidney and mild hepatobiliary disease. Hypertension will eventually affect all children with ARPKD [2, 6].
ARPKD is generally diagnosed by ultrasound in the prenatal period only late during the third trimester of pregnancy [1]. Currently, early and reliable prenatal diagnosis is only possible by molecular genetic analysis. Due to the possibility of misdiagnosis, linkage analysis is no longer a diagnostic method of choice and has been replaced by direct sequencing, usually based on Next Generation Sequencing (NGS) techniques [24].
Neonatal ultrasound features include enlarged echogenic kidneys with poor corticomedullary differentiation, microcysts, or hepatic parenchymal echogenicity. Antenatal ultrasound may show bilaterally enlarged echogenic kidneys, possibly with oligohydramnios, or a small or non-visualized bladder with no urine. If both kidneys are enlarged with diffusely hyperechoic kidneys without cysts, this is highly suggestive of ARPKD (Figs. 3). Older children may present with similar findings, as well as portal hypertension that manifests as splenomegaly and biliary duct dilation [2, 4]. If the parents or grandparents have a history of renal cysts, it may be difficult to distinguish ADPKD from ARPKD in the neonatal period. Microscopic cysts in the neonatal period will usually help to distinguish ARPKD from the macroscopic cysts that develop later in ADPKD [6]. However, with increasing evidence of clinical and genetic overlap between both entities, and the advancement of genetic testing, the gold standard currently is to affirm the correct diagnosis with the use of genetic [1]. Once a preliminary diagnosis of ARPKD is made, US should be performed every 2-3 weeks for serial monitoring of the renal size and amniotic fluid volume of the fetus [24].
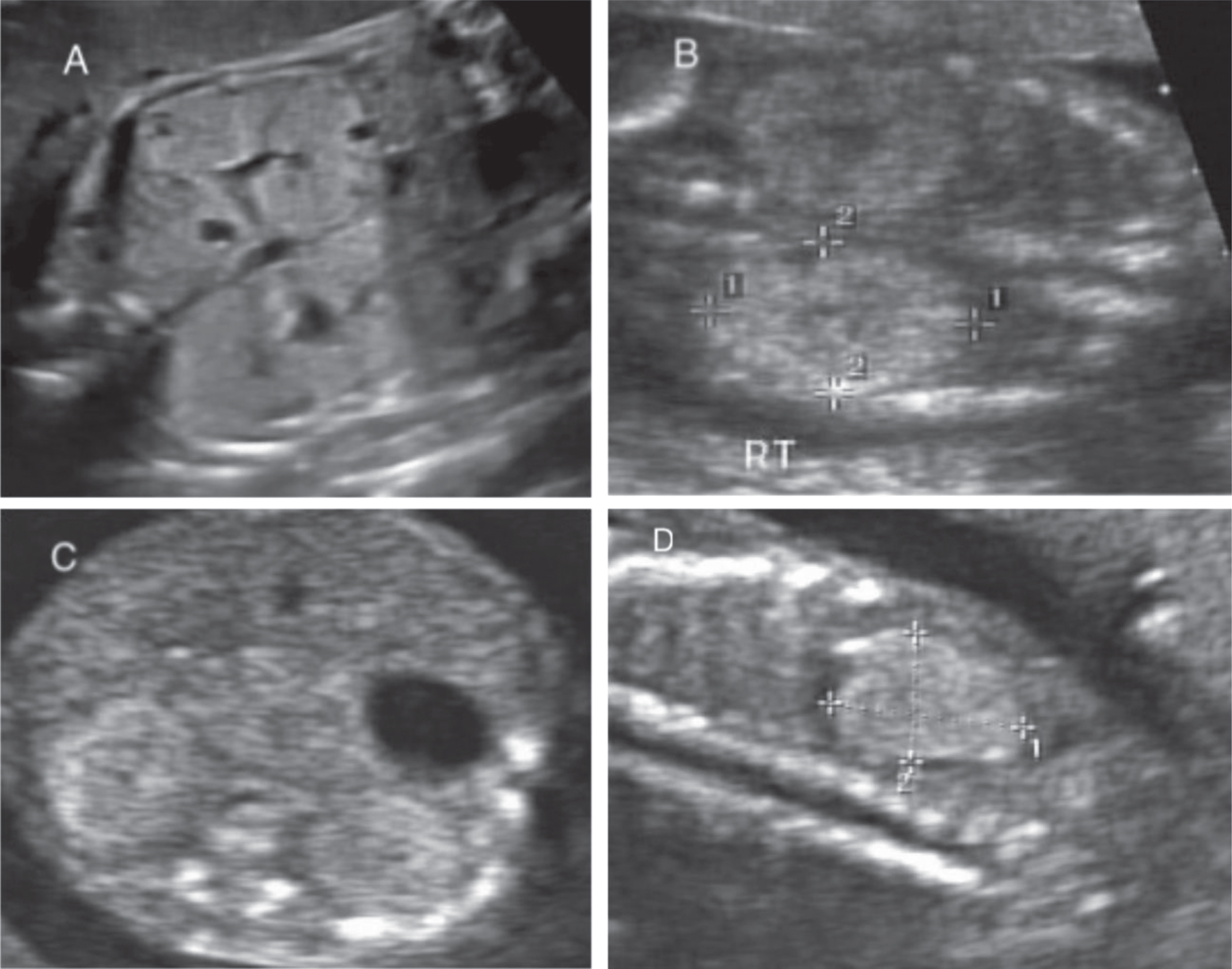
Ultrasound imaging of autosomal recessive polycystic kidney disease (ARPKD). A. Sagittal view of bilateral echogenic kidneys with scattered small cysts. B. Sagittal view of echogenic kidney in ARPKD due to multiple tiny cysts. C. Transverse view of the ARPKD kidneys. D. Sagittal view of echogenic right kidney with small cysts in a fetus with ARPKD.
Systemic evaluations should be done to look for extra-renal anomalies, as other fetal conditions are associated with fetal hyperechogenicity [24]. Other conditions to evaluate for a multitude of other genetic alterations (e.g. Turner syndrome, trisomies 8, 13, 18), hepatorenal fibrocystic disorders such as the peroxisomal Zellweger syndrome, acyl-CoA dehydrogenase defects, or congenital cytomegalovirus (CMV) infection. Amniocentesis should be considered for genetic testing and/or polymerase chain reaction for CMV [24].
Supportive measures should be taken to manage other issues such as electrolyte abnormalities, as these patients are at risk for dehydration. Specifically, hyponatremia may result from impaired urinary dilution, and therefore limiting water intake is recommended without compromising nutrition and by concentrating feeds [1, 24]. Unless there is evidence of hypovolemia, sodium supplementation should be avoided [24].
Urinary tract infections are more likely to present in girls with ARPKD than boys, therefore it is important to rule out structural or functional renal abnormalities. Patients presenting with metabolic acidosis may need bicarbonate replacement [1].
Currently, there is no evidence-based association between ARPKD and intracranial aneurysms, besides some single case reports. Therefore routine screening of asymptomatic patients is not necessary [24].
Infants with ARPKD are at risk for the clinical manifestations of chronic kidney disease, such as anemia, renal osteodystrophy, and cognitive defects as renal function decreases. For patients who develop symptomatic ESRD or portal hypertension, peritoneal dialysis will be the preferred method of renal replacement therapy. The current therapy recommended for controlling blood pressure in these patients is either ACE-I or ARBs. Combination of ACE-I/ARB is not recommended due to increased risk of side effects and no clear added benefit. In the context of CKD, therapy should be aimed at optimizing blood pressure control while minimizing further reduction in GFR [1].
Patients with severe renal or severe hepatobiliary disease should be considered for combined liver-kidney transplant as this may improve their long-term survival and ultimately decrease morbidity and mortality [1].
Hepatic complications remain the primary complication for some ARPKD patients, therefore surgery via endoscopic band ligation or sclerotherapy are recommended if needed. Patients should be annually evaluated for hepatic complications via MRCP, MRI, or ultrasound. Social support is also encouraged for patients and their families as well [1, 3]. Providers should track cognitive, social, and behavioral functioning over time, as well as adherence to medical treatment. To date, there is limited literature available to explicitly direct clinical practice around these issues in ARPKD children [24].
Risk factors for ARPKD patients potentially requiring dialysis within the first year of life are identified as: presence of oligohydramnios or anhydramnios, prenatal kidney enlargement, a low Apgar score, and the need for postnatal breathing support [25]. Caregivers for ARPKD children should maintain a high index of suspicion for cholangitis, and should also be considered in any child with an unexplained fever [24].
Autosomal dominant polycystic kidney disease (ADPKD)
Epidemiology
ADPKD is the most common inherited kidney disease with an incidence of approximately 1:1000 [6]. ADPKD has an autosomal dominant inheritance pattern, but with considerable phenotypic variability. All races and ethnicities are thought to be equally affected by ADPKD. ADPKD is a very rare cause of end-stage renal disease in children, but accounts for 5% of ESRD in adults [6]. Rather than presenting in the neonatal period, affected patients often present in early adulthood, between the ages of 20–40 years old, but it has also been detected in utero by prenatal ultrasound [1].
Genetics and pathophysiology
ADPKD is mainly due to mutations in PKD1 and PKD2, with 80% of gene mutations found in PKD1. PKD1 is a large gene that encodes polycystin 1 (PC1). This membrane-bound protein has receptor-like properties that interact with polycystin 2 (PC2), the product of PKD2. PC1 and PC2 are expressed in many organs such as the kidney, liver, pancreas, heart and the vasculature. PKD2 is a smaller gene and its mutations only account for 15% of mutations in ADPKD. PC2 is a cation channel that modulates the amount of intracellular calcium that regulates downstream signaling processes. ADPKD may also be associated with features of tuberous sclerosis, as the causative gene for tuberous sclerosis, TSC2, lies adjacent to the PKD1 gene on chromosome 16. Patients with a contiguous gene deletion of this region will have tuberous sclerosis and severe polycystic kidney disease usually presenting in utero or diagnosed during infancy. In addition to this genotype-phenotype correlation, mutations in PKD1 and especially those of truncating character usually cause more severe disease than PKD2 mutations, as cysts tend to develop earlier in life. Early manifestations of ADPKD may result in perinatal death [25]. Variable disease expression even within the same family is exemplified by the following pedigree in which perinatal deaths allegedly due to ARPKD occurred in the second and third pregnancy. However, ultrasound screening in other family members revealed bilateral renal cysts in the father (32 years old), paternal grandmother and paternal great-grandmother (80 years old) who invariably showed no or only very mild clinical symptoms. Genetic analysis revealed a PKD2 frameshift mutation in this pedigree, but early and severe manifestation of ADPKD highly suggests a common familial modifying background in line with a gene dosage theory, as recently demonstrated for other families [21, 26]. Various factors have been shown to affect the severity of cyst formation in ADPKD, such as the timing of PKD1 inactivation and the influence of one cyst on neighboring nephrons to create a “snowball effect,” which causes cysts to develop in adjacent tubules [1].
Presentation and diagnosis
ADPKD presents with cystic lesions in the liver, spleen, and pancreas. In addition to these cystic lesions, patients may present with ascular abnormalities, such as mitral valve prolapse and intracranial “berry” aneurysms, although intracranial aneurysms are much less likely to present (Table 1). While these extra-renal manifestations will present much differently in kids than they will in adults, renal findings will present similarly in both age groups of patients. Patients can present in utero with renal cysts. Neonates, children, and adolescents may present with bilaterally enlarged cystic kidneys, left ventricular hypertrophy with or without systemic hypertension, proteinuria, gross hematuria, nephrolithiasis, flank pain, or impaired renal function. It is important to screen newborns with ADPKD for hypertension, as hypertension has been shown to correlate with left ventricular size in these patients. The degree of impaired renal concentrating ability may correlate with disease severity in children with ADPKD. Mitral valve prolapse and extrarenal cysts are less common in pediatric populations in comparison to adult patients [1]. The most cost-effective and least invasive imaging modality for these patients is ultrasound. Ultrasound scan shows enlarged glomerulocystic kidneys with hyperechoic parenchyma, with or without diminished corticomedullary differentiation, and multiple tiny, and possibly some larger and more prominent cysts (Fig. 4). However, as emphasized, ADPKD can phenocopy ARPKD. Both prenatal ultrasounds and family history are vital to properly evaluating the disease. It may be difficult to distinguish ADPKD renal cysts from ARPKD renal cysts in newborns. ADPKD cysts may detach from the tubule and not remain connected to the urinary system, while renal cysts in ARPKD may remain connected within the nephron of origin. ADPKD may become more clinically symptomatic or may remain asymptomatic until adulthood [3]. Table 2 references specific indications from Bergmann et al. for when to initiate genetic testing for patients [21].
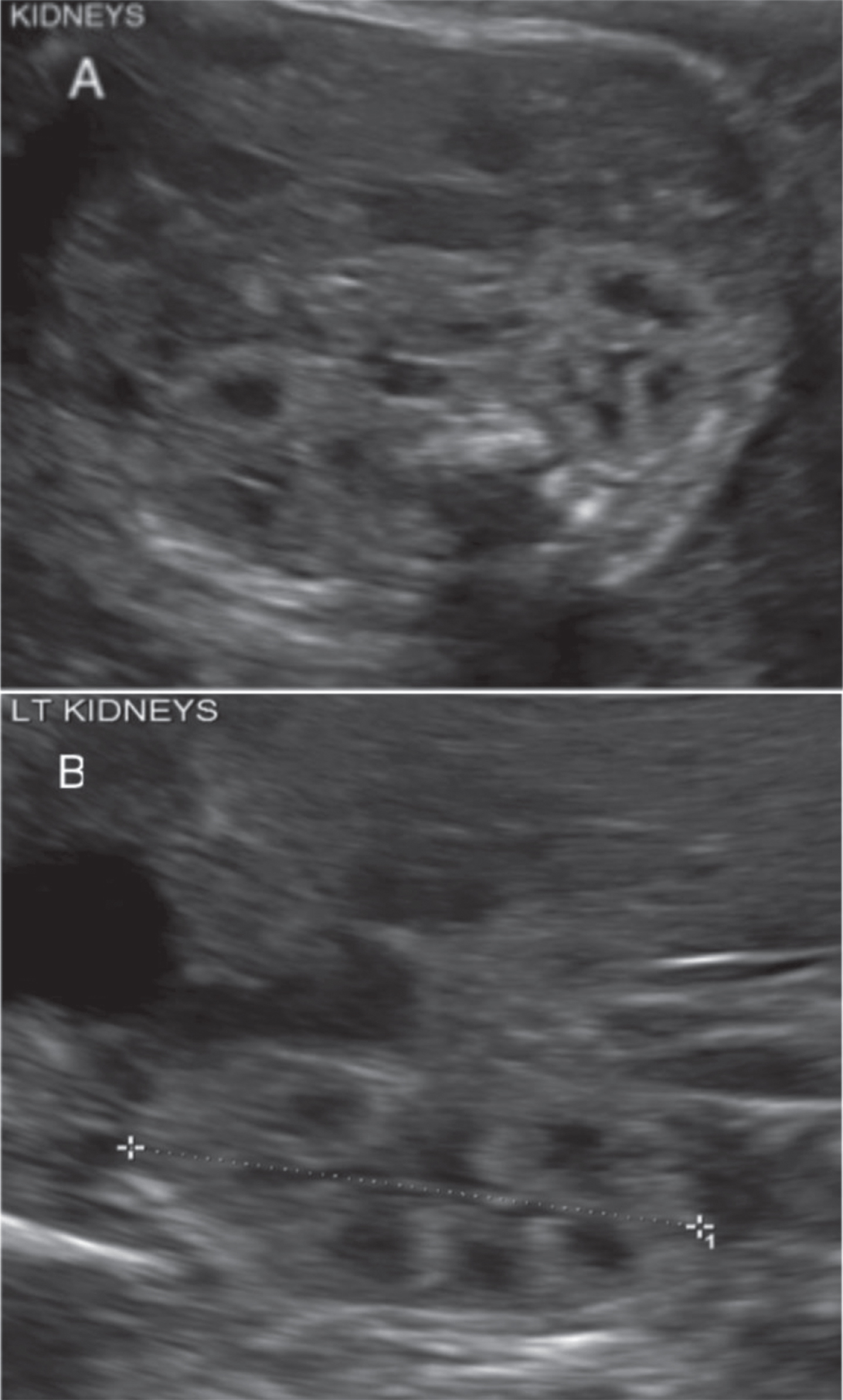
Ultrasound imaging of autosomal dominant polycystic kidney disease (ADPKD). A. Enlarged glomerulocystic kidney with hyperechoic parenchyma, without corticomedullary differentiation, and multiple tiny cysts. B. Markedly enlarged left kidney with multiple cysts.
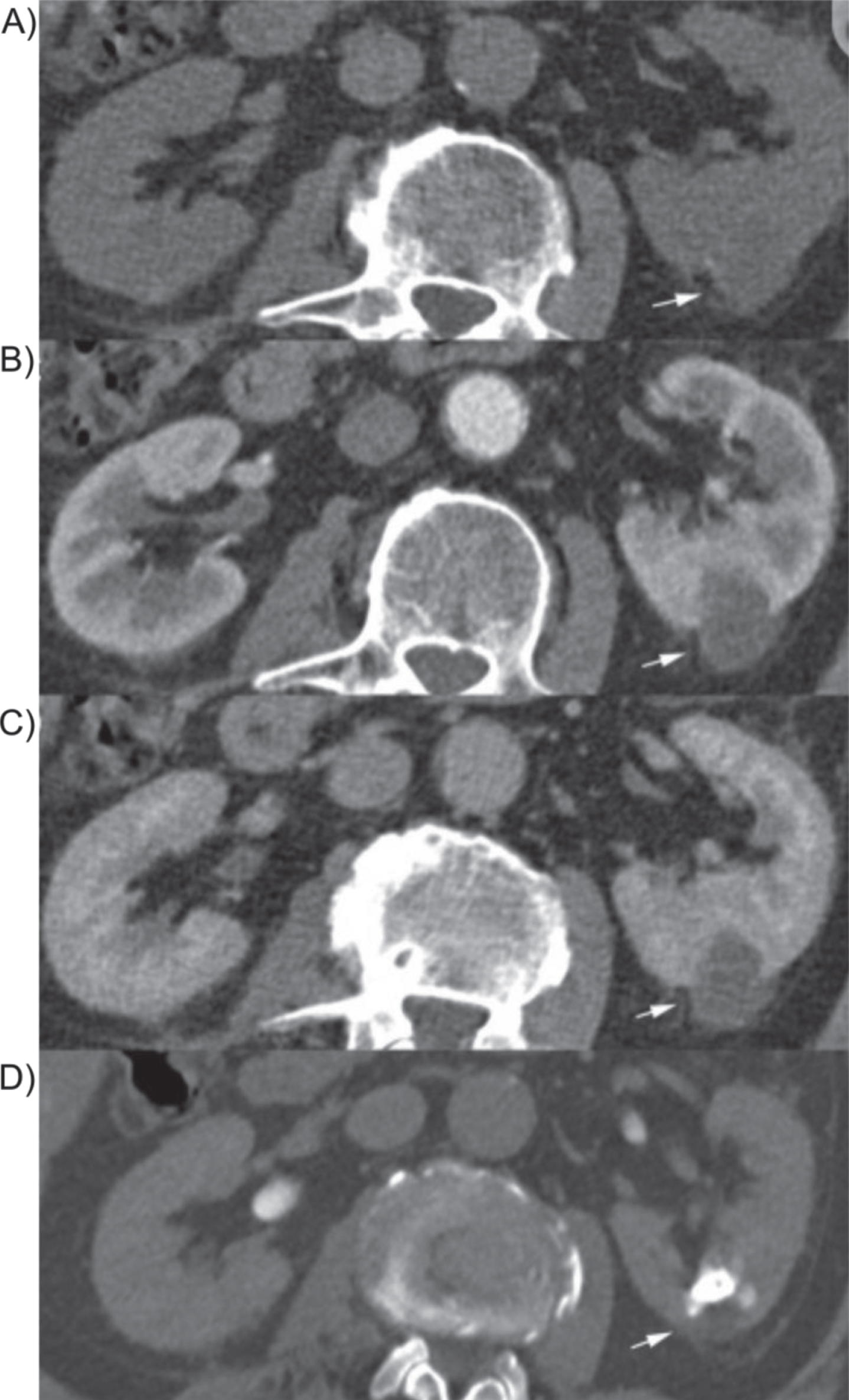
Multiphase CT image of cystic lesion. A multiphase CT including excretion phase was performed. A) Shows a non-contrast CT image, B) shows the lesion during the arterial phase, C) shows the lesion during venous phase and D) during the excretion phase CT 15 min after intravenous contrast administration of the lesion. This lesion is representative of a benign calyceal diverticula.
Indications for genetic testing in ADPKD
ADPKD, autosomal dominant polycystic kidney disease; PKD, polycystic kidney disease.
ADPKD patients need treatment for complications of the natural disease process, as there are limited therapeutic options to halt the progression of the disease. Current recommendations suggest achieving a goal for a maximum blood pressure in 75th percentile in children with ADPKD [27].
Pravastatin has been shown to slow progression of structural kidney disease, and reduces LVH in older children and young adults [1]. No formal consensus has been reached on the use of statins in ADPKD patients [27].
Based on the results of the TEMPO 3:4 trial and REPRISE study, the only FDA-approved, disease-modifying option for adult patients with rapid progression of ADPKD is the selective vasopressin V2 receptor antagonist, tolvaptan. While tolvaptan slows down disease progression, it is not curative and may be associated with adverse effects such as thirst, polyuria, nocturia, polydipsia, and rarely, a significant elevation of liver enzyme levels [28]. However, tolvaptan has been reported as a possible effective treatment for severe neonatal PKD. A female infant diagnosed with ADPKD who presented with massive renal enlargement, respiratory failure, and hyponatremia was started on tolvaptan at one month of age and had resolution of hyponatremia without increase in kidney size by 18 months of age [22].
mTOR inhibitors have been demonstrated to decrease PKD progression in rodent models, but are ineffective in human subjects [29]. Studies demonstrated that somatostatin analogues, such as lantreotide, seem to have a role in ADPKD-associated polycystic liver disease, but did not preserve renal function [30].
Certain medications currently being tested in adults may be tested in pediatric populations in the future, depending on their efficacy and safety in adults: vasopressin V2 receptor antagonist, lixivaptan; AMPK activator, metformin; aldosterone antagonist, spironolactone; and tyrosine receptor kinase inhibitor, tesevatnib [28].
Older patients with flank pain due to an increasing number of renal cysts require pain management. Patients older than age 20 years with cerebral aneurysms or with a positive family history should be closely monitored for symptoms and bleeding [2, 4].
Currently, there are no well-validated measures and predictors for disease progression in children. There is a need to define uniform outcome parameters in this population of pediatric patients to establish the optimal therapeutic regimen [28].
Ultimately, treatment initiation in children with ADPKD will only be feasible with medications that are incentivized by minimal adverse effects, as ADPKD may not lead to obvious clinical symptoms until later in life.
Conclusion
There has been increased understanding in the diagnosis and treatment of genetic and sporadic renal cystic diseases in the neonatal and perinatal populations. Patients diagnosed with ARPKD are able to survive much longer due to advances in neonatal critical care and renal replacement therapy. Proactively treating the complications of ARPKD, such as portal hypertension, has allowed patients to have enhanced quality of life. If eligible, children may receive renal and liver transplants positively altering their life trajectory. Families now have the option to explore pre-implantation genetic diagnosis. Patients with ADPKD should have their blood pressure well controlled to reduce the risk of LVH. For curative measures, it is likely that potential future therapies will utilize multiple agents and modalities, targeting specific changes at different stages of the disease 1. Patients with MCDK require close monitoring and management of proteinuria, hypertension, and renal failure. Clinicians should be aware of first and second line management of the CD depending on the size, shape, and location of diverticula.
Funding
CB is an employee of Limbach and holds a part-time faculty appointment at the University of Freiburg. His research lab receives support from the Deutsche Forschungsgemeinschaft (DFG) DFG BE 3910/8-1 and DFG BE 3910/9-1, the Collaborative Research Centre (SFB) KIDGEM 1140 and from the Federal Ministry of Education and Research (BMBF, 01GM1903I and 01GM1903G).
Conflicts of interest
The authors have no conflicts of interest to disclose.