
Abstract
INTRODUCTION:
Hereditary myosin myopathies are muscle disorders caused by mutations in myosin heavy chain genes. The MYH2 gene encodes the fast 2A skeletal muscle isoform, and mutations manifest as joint contractures, muscle weakness, and external ophthalmoplegia. Muscle biopsy shows decreased type 2A fibers, and vacuoles are sometimes present in adults with progressive disease.
PRESENTATION OF CASE:
This case describes a full term baby boy with hypotonia, dysmorphic features, dysphagia, and aspiration. Whole genome sequencing detected a novel heterozygous variant in the MYH2 gene. Muscle biopsy showed decreased type 2A fibers and vacuoles in myofibers.
DISCUSSION:
Hypotonia and dysphagia are common in infants with a MYH2 myopathy. However, dysmorphic features and vacuoles on biopsy have not previous been described in infants with MYH2 myopathies.
CONCLUSION:
This case reports an unusual phenotype of a rare neonatal-onset congenital myopathy associated with a novel heterozygous variant in MYH2.
Keywords
Introduction
Hereditary myosin myopathies are a heterogeneous group of muscle disorders caused by mutations in myosin heavy chain genes. The various isoforms of myosin determine the physiologic properties of different muscle fiber types, including skeletal muscle fibers. Mutations in developmental isoforms of skeletal myosin, encoded by the MYH3 and MYH8 genes, cause congenital joint contractures and deformities [1]. The myosin 1 isoform, encoded by the MYH7 gene, is expressed in slow type 1 skeletal muscle fibers and cardiac muscle [1, 2]. Mutations in MYH7 cause various myopathies, including myosin storage myopathy and Laing distal myopathy, and are associated with familial hypertrophic and dilated cardiomyopathies [1, 2]. Mutations of the MYH7 gene have also been associated with a central core myopathy [3, 4]. Clinically, these diseases are characterized by distal, axial, and quadricep weakness and hypertrophy of the calves, and muscle biopsy may show minicores, eccentric cores, and fatty infiltration [3, 4].
The myosin 2A isoform is encoded by the MYH2 gene and is expressed in fast type 2 skeletal muscle fibers [1, 2]. Both autosomal dominant and recessive mutations cause a spectrum of diseases that vary in age of presentation and severity of symptoms [1, 2]. Patients presenting at birth have symptoms ranging from mild joint contractures and weakness to more severe hypotonia, dysphagia and ophthalmoplegia [5–10]. Muscle biopsy shows variation in fiber size, disorganized myofilaments, and decreased type 2A fibers [5–10]. Creatine kinase levels are typically normal [6, 8]. Patients presenting with symptoms during childhood, adolescence or adulthood develop progressive proximal muscle weakness and ophthalmoplegia with variable involvement of neck and facial muscles [5, 11–16]. In addition to lacking type 2A fibers, some adults with a progressive course have rimmed vacuoles on muscle biopsy and elevated serum creatine kinase levels [5, 14].
This case describes an infant male who presented with hypotonia, dysmorphic features and dysphagia with aspiration at birth. Whole genome sequencing detected a novel heterozygous mutation in the MYH2 gene in both baby and mother. Muscle biopsy showed decreased type 2A fibers, and ultrastructural studies revealed numerous vacuoles. Vacuoles have been reported in adults with progressive symptoms, but to the best of our knowledge there are no previous reports of vacuoles within the muscles of infants presenting with a MYH2 myopathy. This case describes an infant who present with a MYH2 hereditary myosin myopathy in the neonatal period.
Presentation of case
This is a case of an infant boy born at 37 weeks to a 43-year-old G4P2 mother with past medical history of obesity and type 2 diabetes. Mom had gastric bypass surgery resulting in weight loss and resolution of the type 2 diabetes. She received good prenatal care and pregnancy was complicated by gestational diabetes controlled with diet and exercise. Maternal Group B Streptococcus screen was positive, prenatal labs were otherwise normal. Mom took prenatal vitamins during pregnancy, no other medications or substance use.
Birth history was significant for elective repeat cesarean for oligohydramnios and breech presentation. No maternal antibiotics were given for Group B Streptococcus as there was no labor and membranes were artificially ruptured at the time of delivery with clear fluid. Baby’s birth weight was 2725 grams and APGAR scores were 8/9. Dad had one episode of fever and mom had a cough at the time of delivery. This was early in the COVID-19 pandemic and testing was not available for parents, however baby subsequently tested negative.
Baby was born at an outside facility and was transferred to our NICU at 7 hours of life. Physical exam was notable for micrognathia, a high arched palate, a prominent occipital protuberance, and hypospadias. Peripheral tone was decreased, and central tone was intact (Fig. 1). Reflexes and coordination were intact throughout, and baby withdrew to pain in all extremities. Mom notably had a small lower jaw and reported being a “floppy baby” and sometimes has “difficulty swallowing.” Baby had poor feeding efforts and nasogastric tube was placed day 1 of life to supplement oral feeds. He tolerated formula feeds (Similac Pro Advance 20 kcal/oz) with appropriate weight gain.
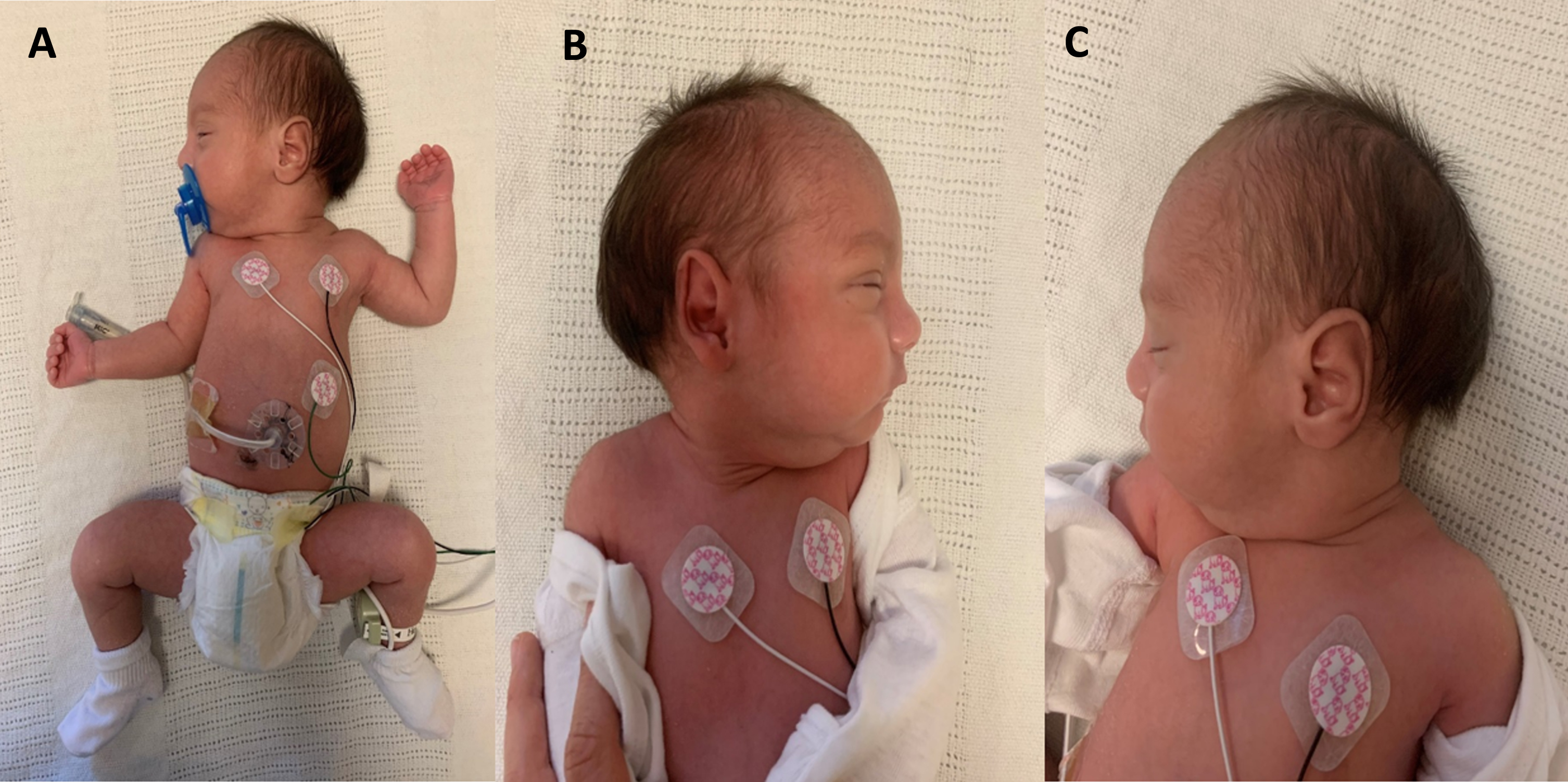
Dysmorphic feature and hypotonia. 20 day old neonate born at 37 weeks. Physical exam shows dysmorphic facial features: flat facies, depressed nasal bridge, high arched palate, micrognathia and retrognathia. Other abnormal physical exam findings include prominent occipital protuberance, peripheral hypotonia of both upper and lower extremities with intact reflexes and hypospadias. G-tube in place as baby had aspiration on fluoroscopic swallow study. A: picture of whole infant. B: prominent facial features. C: prominent occipital protuberance.
CBC and CMP were unremarkable. Blood cultures were negative, and antibiotics were never initiated. On day 3 of life, brain MRI showed myelination appropriate for age with normal appearing structures and incidental finding of cavum septum pellucidum, a normal anatomical variation (Fig. 2). Metabolic work up included normal serum ammonia (77 umol/L; normal newborn < 100 umol/L) and pyruvate (0.033 mmol/L; normal 0.030–0.107). Serum amino acid and urine organic acid profiles showed no evidence of a metabolic disorder. Serum creatine kinase (57 U/L; normal < 182 U/L) and liver function tests were normal. No abnormalities were detected on chromosomal microarray. First and second newborn screenings on day 0 and 5 of life showed normal findings.
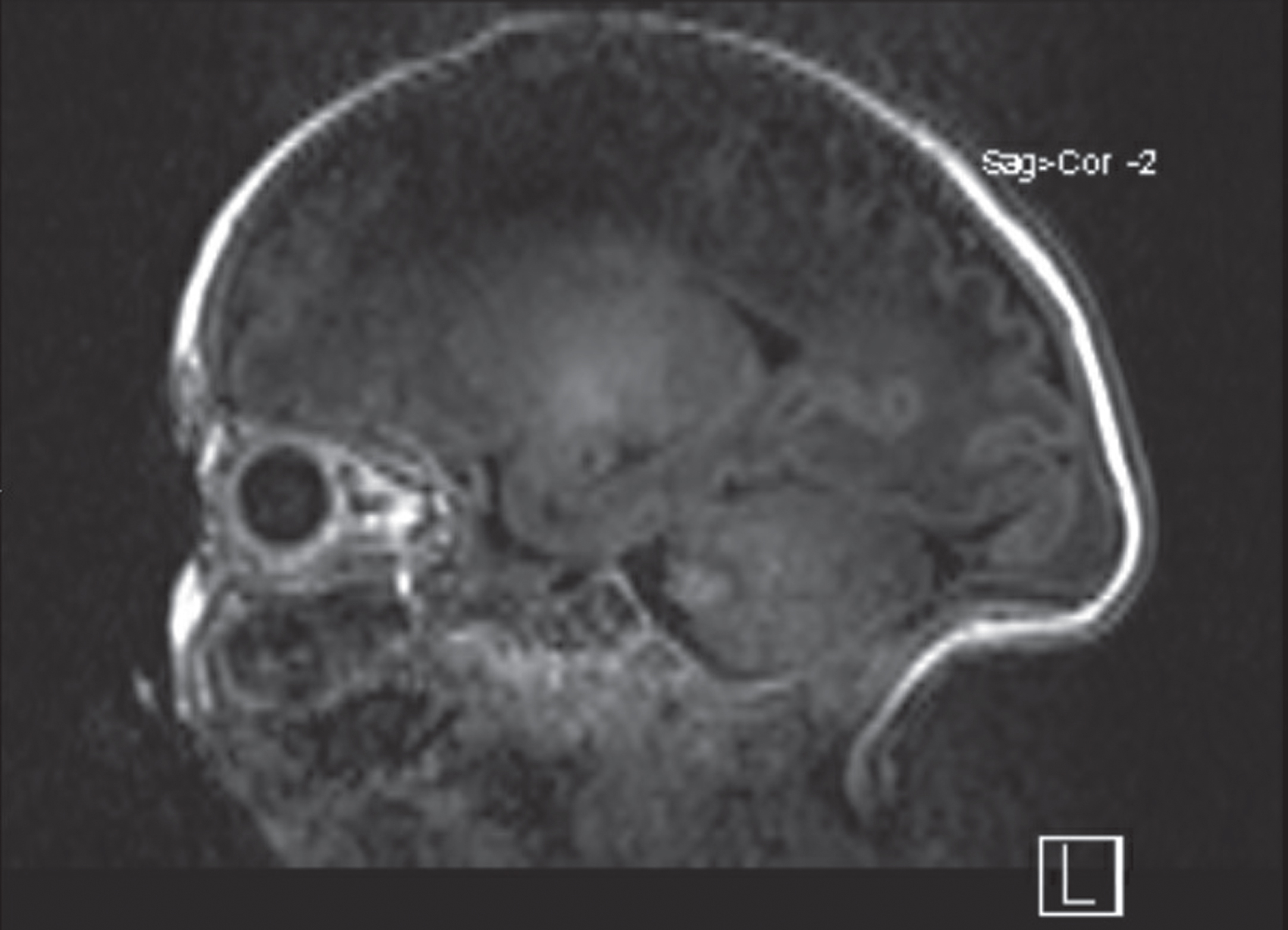
Brain MRI. Noncontrast saggital view of Brain MRI is unremarkable. Myelination, sulcation and gyration is appropriate for age. Prominent occipital protruberance and cavum of the septum pellucidum.
On day 4 of life, he had episodes of oxygen desaturation with all attempted bottle feeds. Video fluoroscopic swallow study on day 5 of life showed impairment in both the oral and pharyngeal phases of swallowing with penetration and aspiration of thin and thickened liquids. All future feeds were exclusively given thorough the nasogastric tube. Despite over a week of suck and swallow therapy with occupational therapy, repeat video fluoroscopic swallow study on day 16 of life showed continued penetration and aspiration of all liquids. On day 19 of life G-tube was placed and muscle biopsy was performed. A sample of baby’s right quadriceps was sent to a specialized neuromuscular pathology department for evaluation. On day 22 of life blood samples were obtained from baby and both parents for whole genome sequencing. Baby had an uneventful post-surgical course and was discharged home from the NICU on day 23 of life.
Whole genome sequencing was performed using next generation sequencing technology. Results were received on day of life 37 and were significant for a heterozygous c.3709C > T (p.Leu1237Phe) variant in the MYH2 gene. This sequence change replaces leucine with phenylalanine at codon 1237 of the MYH2 protein (p.Leu1237Phe) and is located on chromosome 17p13.1
The muscle biopsy sample was examined with light and electron microscopy (Fig. 3). Hematoxylin and Eosin staining showed minimal variation in fiber size with normal diameter of fiber size for age (12–14 um) (Fig. 3A.). There was no evidence of acute or chronic myopathic features at light microscopy. Fiber types were identified with ATPases, slow myosin, fast myosin and toluidine blue revealing both type 1 and type 2 fibers. The number of type 2A fibers was decreased (light brown) compared to type 2B fibers (pink fibers) (Fig. 3C).
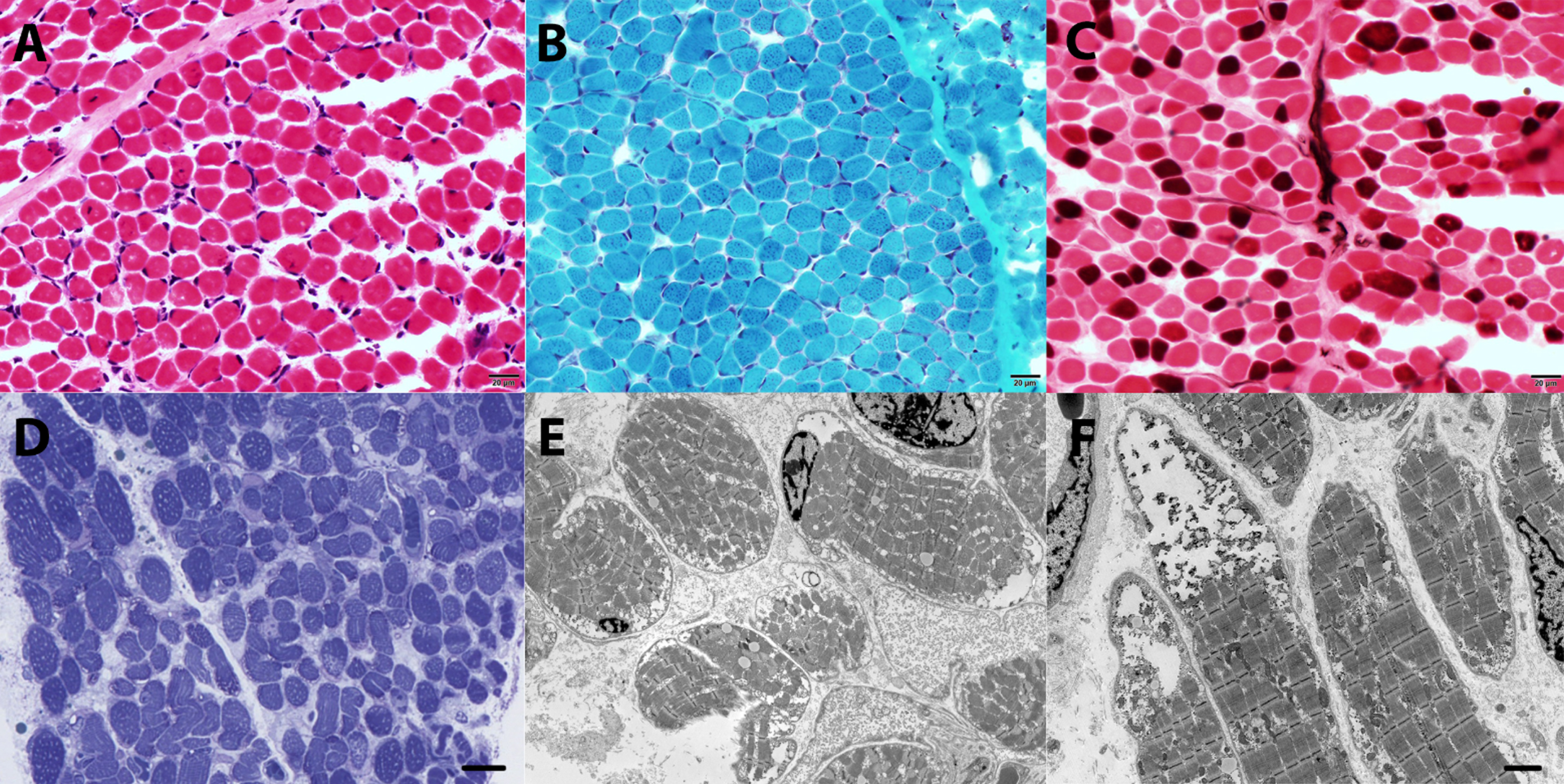
Muscle biopsy, quadriceps. A. Hematoxylin and Eosin (H&E) staining (standard thickness 8 micros) shows minimal variation in fiber size, with normal size for age, normally placed nuclei at the periphery, no evidence of vacuoles, fibrosis, fat replacement or myopathic changes. B. Modified Gomori Trichrome staining is within normal limits with no evidence of rimmed vacuoles, abnormal myofibrillar distribution or endomysial fibrosis. C. ATPases pH 4.6 shows a type 2B predominant muscle (pink fibers), with a paucity of type 2A fibers (light brown). D. Toluidine blue staining for semi-thin (1 micron) epon embedded section shows several fibers with subsarcolemmal vacuoles that were not evident in conventional light microscopy. E. Electron microscopy, cross sections show several fibers with subsarcolemmal and intermyofibrillary vacuoles. Some vacuoles are empty, while others have granular non-membrane bound deposits. F. Electron microscopy, longitudinal sections show large subasrcolemmal vacuoles with electro dense glycogen-like deposits. Scale bar A-D 20 micrometers; E & F: 5 micrometers.
There was no evidence of fiber atrophy, hypotrophy, or hypertrophy. Oxidative enzymes (NADH, SDH and COX) were within normal limits. Modified gomori trichrome was unremarkable with no evidence of rimmed vacuoles or abnormal myofibrillar arrangement (Fig. 3B). A limited metabolic panel that included PAS, Oil red O, myodenylate deaminase, acid phosphatase, and phosphofructokinase was unremarkable.
Toluidine blue staining for semi-thin (1 micron) epon embedded section shows several fibers with subsarcolemmal vacuoles that were not evident in conventional light microscopy (Fig. 3D). Electron microscopy showed numerous vacuoles in several fibers (Fig. 3E, 3F). The content of the vacuoles varied from empty to electro-lucid material and granular glycogen-like particles. Occasional lysosomal vacuoles were visualized. Rare fibers with disorganized filaments were identified but were not a predominant feature. There were mildly increased lipid droplets. Cores, minicores or rods were not identified. The mitochondria were within normal limits and had normal internal architecture.
Baby was 5 months old at his most recent clinic visit. He was not meeting gross motor milestones such as sitting with support or lifting his chest off the ground when placed prone. He had some fine motor control and was babbling and meeting social milestones. He was taking some table foods by mouth, but all liquids continue to be given through his G-tube. Baby is pending evaluation by a neuromuscular specialist and geneticist. He was hospitalized at 2 months old for pyelonephritis. He follows with urology; plan is for future surgical repair of hypospadias.
This is a case of a full term infant male with hypotonia, dysmorphic features, and dysphagia with aspiration requiring G-tube feeds. Initial workup including brain MRI, metabolic studies, creatine kinase and microarray was unrevealing. Whole genome sequencing showed a novel heterozygous variant in the MYH2 gene associated with hereditary myosin myopathy. Muscle biopsy showed decreased type 2A fibers, and vacuoles were seen on electron microscopy. This patient’s early diagnosis presented the rare opportunity to further characterize the pathological findings associated with neonatal-onset MYH2 hereditary myosin myopathies.
Hypotonia and dysphagia with normal creatine kinase levels are consistent findings in symptomatic infants with MYH2 myopathies [5–10]. In a similar case, an infant with an autosomal dominant MYH2 mutation presented with hypotonia and dysphagia with aspiration requiring G-tube feeds [6]. Two brothers with chronic dysphagia since birth, one of whom required partial pneumonectomy secondary to severe aspiration, were homozygous for autosomal recessive mutations in the MYH2 gene [7]. In another family, two infants with recessive MYH2 gene mutations were slow to feed with frequent choking [8].
Our patient also had a high arched palate, micrognathia, a prominent occipital protuberance and hypospadias. We identified one previous report of an infant with a MYH2 myopathy and an arched palate [6]. However, to our knowledge our patient’s additional features have not been described in MYH2 mutations. Baby’s mother, who was heterozygous for the MYH2 mutation, also had a small lower jaw. The micrognathia may be associated with this variant as mom also reports being a “floppy baby” and sometimes has “difficulty swallowing and chocking episodes.” The prominent occipital protuberance might not be related to the MYH2 mutation, although no other genetic conditions were identified on whole genome sequencing. Lastly, hypospadias is common in typically developing newborns and may not be associated with this MYH2 mutation.
Whole genome sequencing was significant for a heterozygous c.3709C > T (p.Leu1237Phe) variant in the MYH2 gene. This sequence change replaces leucine with phenylalanine at codon 1237 of the MYH2 protein (p.Leu1237Phe) and is located on chromosome 17p13.1. The leucine residue is highly conserved and there is a small physicochemical difference between leucine and phenylalanine. This variant is present in population databases (rs138265883, ExAC 0.009%). This variant has not been reported in the literature in individuals with MYH2-related disease. ClinVar contains an entry for this variant (Variation ID: 321672). Algorithms developed to predict the effect of missense changes on protein structure and function are either unavailable or do not agree on the potential impact of this missense change. So far, the available evidence is currently insufficient to determine the role of this variant in disease. Therefore, it has been classified as a variant of uncertain significance. Mom was found to be heterozygous for the variant while dad was negative [17]. This variant had yet to be functionally characterized in the literature [17].
Muscle biopsies from patients with MYH2 myopathies typically show disorganized fibers that vary in size with few or absent type 2A fibers [2, 18]. Our patient had minimal disorganized fibers and variation in fiber size was typical for age, however, the number of type 2A fibers were decreased. Vacuoles were also visualized on electron microscopy. To the best of our knowledge, there are no previous reports of vacuoles in the myofibers of symptomatic infants with a confirmed MYH2 myopathy. However, of these cases only one reports analyzing the ultrastructural components of muscle in symptomatic infants [8]. Storage diseases are a more common cause of vacuolar myopathies in infants, some of which can present with similar symptoms of muscle weakness and hypotonia. Our patient had an unrevealing metabolic workup, and no additional genetic mutations were discovered on whole genome sequencing making a storage disease less likely.
Though not previously described in infants, rimmed vacuoles are well documented in a subset of adults with progressive symptoms and elevated creatine kinase levels [5, 14]. The vacuoles are visible without the use of ultrastructural studies and may be related to chronicity, only appearing overtime with more advanced disease [5, 14]. In the case of one patient, vacuoles were visualized only under electron microscopy at the age of 17 years old. However, when the patient had more profound symptoms at 39 years of age, vacuoles were seen at light microscopy [13]. If our patient follows a more progressive course, vacuoles may eventually be evident at light microscopy.
Conclusion
Hereditary myosin myopathies are a broad spectrum of diseases that have only recently been described. Our patient presented with typical findings of a neonatal-onset MYH2 myopathy including hypotonia, dysphagia, and a normal creatine kinase level. However, this baby also had dysmorphic features that have not previously been described in MYH2 myopathies. Muscle biopsy showed decreased type 2A muscle fibers, and vacuoles were visualized on electron microscopy. Rimmed vacuoles are well described in a subset of adult patients with delayed onset of progressive symptoms and elevated creatine kinase levels. To the best of our knowledge, this is the first report of vacuoles in the myofibers of infants with MYH2 myopathy presenting with symptoms in the neonatal period.
Consent
Written consent for publication and photography was obtained and signed by both parents.
Footnotes
Acknowledgments
Not applicable.
Funding
Not applicable.
Ethical approval
Institutional ethics committee approval was taken for the publication.
Conflicts of interests
All authors declare no conflict of interests or disclosures.