
Abstract
Background:
The central nervous system involvement, in terms of a maladaptive sensory-motor plasticity, is well known in patients with dystrophic myotonias (DMs). To date, there are no data suggesting a central nervous system involvement in non-dystrophic myotonias (NDMs).
Objective:
To investigate sensory-motor plasticity in patients with Myotonia Congenita (MC) and Paramyotonia Congenita (PMC) with or without mexiletine.
Methods:
Twelve patients with a clinical, genetic, and electromyographic evidence of MC, fifteen with PMC, and 25 healthy controls (HC) were included in the study. TMS on both primary motor cortices (M1) and a rapid paired associative stimulation (rPAS) paradigm were carried out to assess M1 excitability and sensory-motor plasticity.
Results:
patients showed a higher cortical excitability and a deterioration of the topographic specificity of rPAS aftereffects, as compared to HCs. There was no correlation among neurophysiological and clinical-demographic characteristics. Noteworthy, the patients who were under mexiletine showed a minor impairment of the topographic specificity of rPAS aftereffects as compared to those who did not take the drug.
Conclusion:
our findings could suggest the deterioration of cortical sensory-motor plasticity in patients with NDMs as a trait of the disease.
Keywords
Introduction
Myotonia is the difficulty to relax muscles after maximum voluntary contraction, and is a common symptom among different neuromuscular disorders, with variable severity and with or without associated symptoms (Sansone et al., 2016).
Typically, myotonias are classified in: (i) dystrophic (DMs), including myotonic dystrophy type 1 (DM1) and type 2 (DM2), related to mutations in myotonic dystrophy protein kinase (DMPK) gene and nucleic acid-binding protein (CNBP) gene (previously known as zinc finger 9 gene, ZNF9), respectively; and (ii) non-dystrophic (NDMs), including Myotonia Congenita (MC) and Paramyotonia Congenita (PMC), which are due to mutations in the voltage-gated ion channels CLCN1 and SCN4A, respectively (Heatwole et al., 2013; Meola & Cardani, 2015; Matthews et al., 2010). Such ion-channel mutations determine an abnormal fast inactivation, a shift in activation, or an altered slow inactivation of the channel. Overall, these dysfunctions lead to persistent late Na + currents (i.e., prolonged depolarization), causing repetitive discharges (in myotonia) or paralysis (in periodic paralyses).
However, myotonia in some disorders is part of a multiorgan impairment, including the brain, which may be related, at least in part, to ion channel or kinase involvement (Sansone et al., 2016; Cardani et al., 2012; Suominen et al., 2008; Sun et al., 2011; Mankodi et al., 2002; Bugiardini et al., 2015). About that, a subclinical impairment of corticomotor system has been demonstrated in patients with DM1 as one of the multisystemic manifestation of the disease (Oliveri et al., 1997). Further, Portaro et al. demonstrated a deterioration of cortical plasticity mechanisms within sensorimotor cortex in patients with DM1, which was independent of the primitive muscle damage (Portaro et al., 2017). Altogether, these findings may indicate that CNS abnormalities in patients with DM1 represent an additional epiphenomenon of a multisystemic syndrome as the DM1 is, given that there was no correlation between neurophysiological parameters and the degree of clinical impairment (Oliveri et al., 1997; Portaro et al., 2017).
Clinical and electrical features of myotonia may be quite similar between DMs and NDMs, yet while patients with DM1 have a brain disorder, this is not the case for NDMs. To the best of our knowledge, there are no data on brain involvement in patients with NDMs. Herein, we investigated corticospinal output and sensorimotor plasticity in patients with NDMs by using Transcranial Magnetic Stimulation (TMS), to define sensorimotor involvement in this condition and the possible relationships with clinical-demographic characteristics. We also investigated whether mexiletine, the drug commonly used as anti-myotonia medication, has effects on potential CNS target.
Materials and methods
Subjects
Twenty patients with a clinical, genetic, and electromyographic evidence of MC, and fifteen with PMC, were included in the study. All patients were evaluated by using the Clinical Rating Scale (CRS) (a 0–4 scale that rates the presence of myotonia/paramyotonia, functional weakness and impairment as it follows: -0- absence of myotonia/paramyotonia; -1- presence of myotonia/paramyotonia and/or mild functional weakness without functional impairment; -2- moderate muscle weakness leading to some degree of functional impairment; -3- muscle weakness with severe functional impairment and in some cases resulting in the subjects being bound to a wheel-chair; and -4- bedridden), and the Myotonia Severity Scale (MSS) (a 0–4 scale that rates the entity of myotonia/paramyotonia as it follows: -0- absence of myotonia/paramyotonia, -1- the minimal, -2- moderate, -3- severe, and -4- the worst myotonia/paramyotonia experienced). Patients had neither clinical nor laboratory evidence of neurocognitive (global cognitive status was assessed using the Addenbrooke’s Cognitive Examination– Revised) (Mioshi et al., 2006), neuropsychological (mood and behavioral disorders were examined using the Hamilton Depression and Anxiety Rating scale) (Hamilton, 1959, 1960), and other neurological disorders, nor diseases affecting the peripheral nervous system, did not take drugs modifying muscles and nerve excitability beyond mexiletine, and had no contraindications to TMS. Experimenters were blinded towards group assignment of participants. Twenty-five heathy controls (HC) were enrolled as a control group (12 females and 13 males; mean age 33±7 years). The study was reviewed and approved by the Local Ethics Committee, and conducted according to the principles expressed in the Declaration of Helsinki. All the subjects gave their written informed consent to study participation.
Experimental sessions
The subjects were seated in a comfortable chair, with both the arms resting on a pillow put over their thighs. The measurements of motor-evoked potential (MEP) and central motor conduction time (CMCT) (i.e., the time taken for the nerve impulse to travel from the motor cortex to the spinal motoneuron) were carried out by using TMS on both primary motor cortices (M1) through a high-power Magstim 200 stimulator (Magstim Company; Whitland, Dyfed, UK) with a 9 cm circular coil centered over the vertex. The current direction in the coil was counter-clockwise or clockwise for preferential activation of the left and right hemisphere, respectively (Rossini et al., 1994). Monophasic TMS pulses were delivered at an intensity that was progressively increased in steps of 10% maximum stimulator output until a reproducible MEP was obtained from tonically active FDI (at approximately 10–15% of maximum force level; audiovisual feedback of ongoing EMG activity was provided to ensure a constant strength). Therefore, 10 MEPs were recorded at the same stimulation intensity and, then, superimposed to identify the earliest onset latency (Walsh, 2004).
CMCT was calculated according to the following formula: CMCT = MEP– (M+F– 1)/2 (normal value <8 ms), i.e., by subtracting the peripheral MCT (PMCT) (i.e., from the spinal motoneuron to the muscle, calculated by (M+F– 1)/2 ms, where M and F are the distal motor and F-wave latencies, respectively, and 1ms is the time to allow for re-excitation of the motoneuron at the level of the spinal cord) from the latency of the MEP (i.e., from motor cortex to muscle) (Walsh, 2004; Koh & Eyre, 1988; Eyre et al., 1991). M- and F-wave latencies were recorded by stimulating the left and right ulnar nerve at the wrist.
The recruitment curve (input-output, I-O, curve) was assessed to estimate cortical excitability. First, we recorded the rectified M-waves by supramaximally stimulating the ulnar nerve at the wrist of the dominant side by means of a pair of surface disk electrodes. The area under the curve of the M-wave was calculated and used as individual reference parameter against which to compare MEPs. Then, we determined the resting motor threshold (RMT) (MacDonnell et al., 1991) from the relaxed first dorsal interosseous muscle (FDI) using a standard figure-of-eight coil wired to a high-power Magstim 200 stimulator. MT was defined for each individual and defined as the minimal intensity of stimulation capable of inducing MEPs greater than 50μV peak-to-peak amplitude in at least 5 out of 10 consecutive trials (Rossini, 1994). This was the reference value to which the intensity of TMS stimulation was set. Then, 10 pulses for 12 different stimulus intensities (RMT±10% steps, from – 40 to +70% ) were randomly delivered, with an interstimulus interval of 10 s, so as to avoid any interference between two successive pulses (Chen et al., 1997). MEPs were rectified and the area under the curve for each response was calculated and averaged across the subjects. Data were normalized to the FDI supramaximal M-wave and submitted to further analysis.
In a separate session, 10 MEPs were recorded using a stimulation intensity of 120% RMT during slight tonic contraction at approximately 10–15% of maximum force level. Audiovisual feedback of ongoing EMG activity was provided to ensure a constant strength. These active trials were used to measure the duration of the cortical silent period (CSP), which is a marker of long-lasting intracortical inhibition (presumably GABAergic) (Siebner et al., 1998; Werhahn et al., 1999). For CSP measurements, EMG traces were rectified but not averaged. The duration of the CSP was measured in each trial and defined as the time from the onset of the MEP to the reappearance of sustained EMG activity (Orth & Rothwell, 2004).
Finally, we assessed sensory-motor plasticity using a rapid paired associative stimulation (rPAS) paradigm (Quartarone et al., 2006). rPAS consisted of 600 pairs of stimuli delivered to the left M1 at a rate of 5 Hz for 2 min. Each pair of stimuli consisted of an electrical conditioning stimulus given to the right ulnar nerve at twice the sensory threshold followed, after 25 ms, by a biphasic transcranial magnetic stimulus (at 90% of active motor threshold -AMT) given to the left M1 by using a Magstim repetitive stimulator wired to a figure-of-eight coil (Quartarone et al., 2006). In this protocol, MEPs were recorded from both FDI and abductor pollicis brevis muscle (APB) of the right hand (using monophasic stimuli delivered through a standard figure-of-eight coil wired to a high-power Magstim 200 stimulator at 120% RMT) before (TPRE) and after (immediately, T0, 30-minute, T30, and 60-minute, T60) the end of rPAS.
Data recording and analysis
EMG signals were recorded with Ag– AgCl surface electrodes from the muscles using a belly-tendon montage. Signals were amplified and bandpass filtered (10 Hz to 1 KHz) by a Digitimer D-150 amplifier and stored at a sampling rate of 10 kHz on a personal computer for off-line analysis (Signal Software; Cambridge Electronic Design, Cambridge, UK).
The baseline neurophysiological data were compared using ANOVA with Bonferroni correction. According to previous works, patient’s values were considered abnormal when they were±2SD of the normal mean (Oliveri et al., 1997).
I-O curve was analyzed by using an ANOVA with the factors group (two levels: NDMs and HC) and stimulation intensity (12 levels). Newman– Keuls test was used as post-hoc test for multiple comparisons.
The effects of rPAS on peak-to-peak MEP amplitude were evaluated by using a repeated measures ANOVA with the factor time (four levels: TPRE, T0, T30, and T60), muscle (two levels: APB and FDI), and group (two levels: NDMs and HC). The factor muscle was employed to assess the topographic specificity of the rPAS aftereffects. Conditional on a significant time×muscle interaction, the factor mexiletine intake (two levels: yes and no) and disease-type (two levels: MC and PMC) were added to the analysis. The Greenhouse-Geisser method was used if necessary to correct for non-sphericity. Conditional on a significant F value, post-hoc t-tests were performed to explore the strength of main effects and the patterns of interaction between experimental factors. A Bonferroni corrected p-value of <0.05 was considered significant. All data are given as mean±SD. Age, gender, age of symptomatology onset, comorbidities, and Myotonia Severity Scale (MSS) score were correlated with I-O curve magnitude of increase and the rPAS topographic specificity of aftereffects by calculating a Spearman correlation test.
Results
All the recruited patients completed the experimental procedure, without reporting any adverse effect during or after the experiment. Given that we explored the possibility of a CNS involvement in the NMDs and that most of the patients were on-treatment with mexiletine, the patient sample was considered as a whole, in that we included both PMC and MC and patients with or without mexiletine. The NDM group consisted of 20 patients with MC (10 females and 10 males, means age 31±9 years, with a disease duration of 6±2 years, CRS of 1-2, MSS of 0–2, and all with mexiletine at a dosage of 400–600 mg/daily), and 15 with PMC (7 females and 8 males, means age 32±5 years, with a disease duration of 18±11 years, CRS and MSS of 0–2; five of them were treated with mexiletine at a dosage of 400–600 mg/daily). There were no significant clinical-demographic differences between patients with or without mexiletine. Both neurocognitive and neuropsychological profile were normal.
There were no significant differences between the groups concerning CMCT (HCs 6.2 ms; NDMs 6.5 ms; p = 0.3) and MEP amplitude (HCs 2 mV; NDMs 2.2 mV; p = 0.3) (Fig. 1A). RMT was lower (although non-significant) in patients than HC (p = 0.4) (Fig. 1A). CSP duration was significantly shorter in NDMs than HCs (group effect F (1,58) = 9.2, p < 0.001) (Fig. 1A). The tonic contraction of the FDI modulated differently MEP amplitude of FDI and APB (group×muscle F (1,58) = 53, p < 0.001), i.e., MEP amplitude increased in FDI and decreased in APB in HCs (p < 0.001), whereas MEP equally increased in both the muscles in patients with NDMs (p = 0.5) (Fig. 1B).
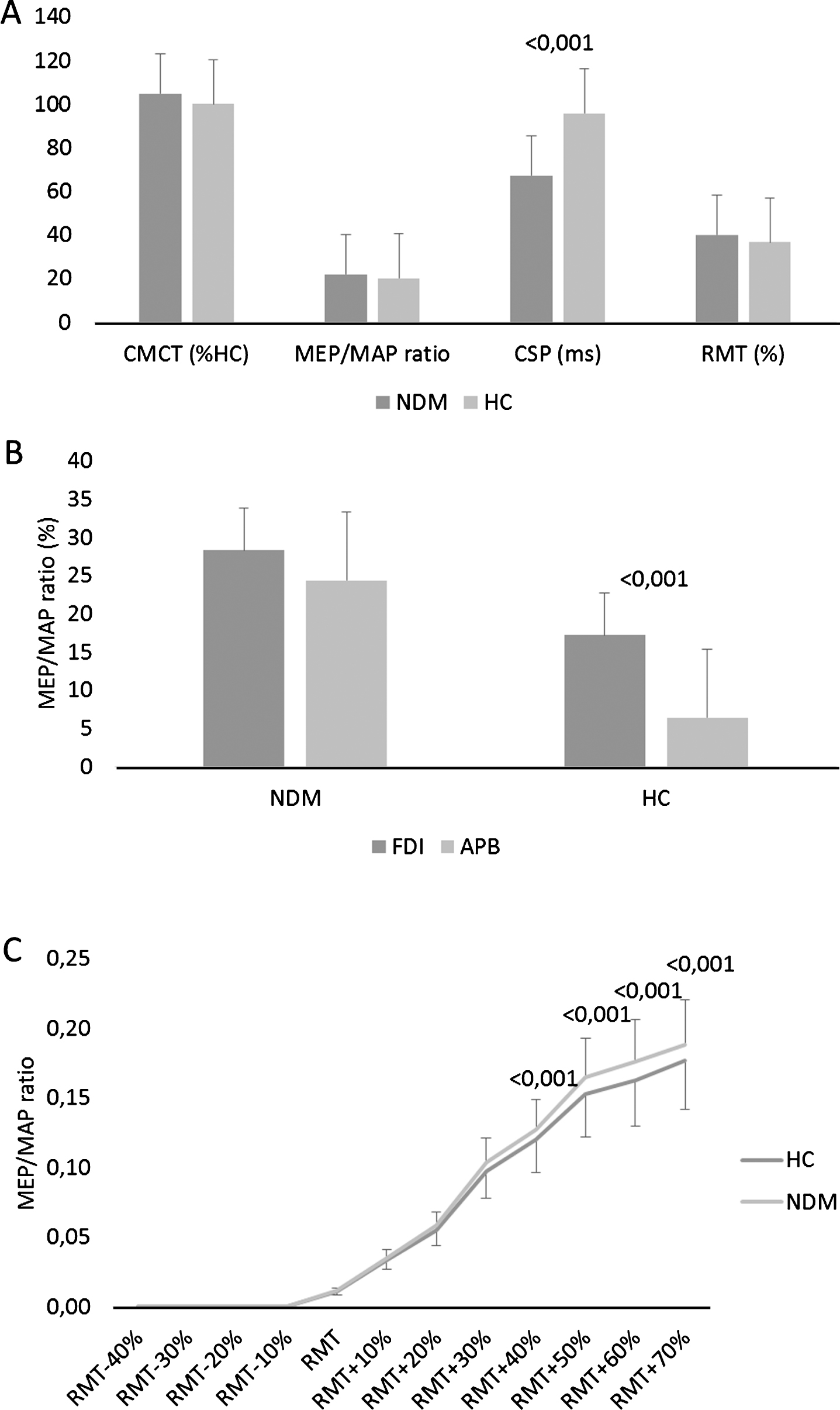
(A) Central motor conduction time (CMCT) as percentage of healthy controls (HC), motor evoked potential/motor action potential (MEP/MAP) ratio, cortical silent period (CSP) duration in ms, and resting motor threshold (RMT) as % of stimulator output in patients with non-dystrophic myotonia (NDM) and HC. (B) MEP/MAP ratio during tonic contraction of first dorsal interosseous (FDI) with resting abductor pollicis brevis (APB). (C) Input– output curves measured from FDI. Each point represents the normalized MEPs averaged across the patients (area under the curve) for each intensity of stimulation. Stimulation intensities refer to the individual RMT expressed in percentage. MEPs were rectified and the area under the curve for each response was calculated. Data were normalized to the FDI supramaximal M-wave. *refer to significant changes as compared to baseline (p < 0.05). Vertical error bars refer to SD.
Statistical analysis of the overall I-O curve data disclosed a significant group×stimulation-intensity interaction (F (11,638) = 14, p < 0.001), i.e., the patients with NDMs showed a significantly higher stimulation-intensity effect than HC did (Fig. 1C).
About rPAS aftereffects, there were significant temporal and topographic differences concerning MEP amplitude increase following rPAS application (time×group×muscle F (3,174) = 3.6, p = 0.01) (Fig. 2). In particular, a topographically specific MEP amplitude increase was detectable in HCs (time×muscle F (3,72) = 4.1, p = 0.007; muscle F (1,24) = 8.1, p = 0.001; FDI time F (3,72) = 9.7, p < 0.001; APB time p = 0.5) but not in patients (time×muscle p = 0.5).
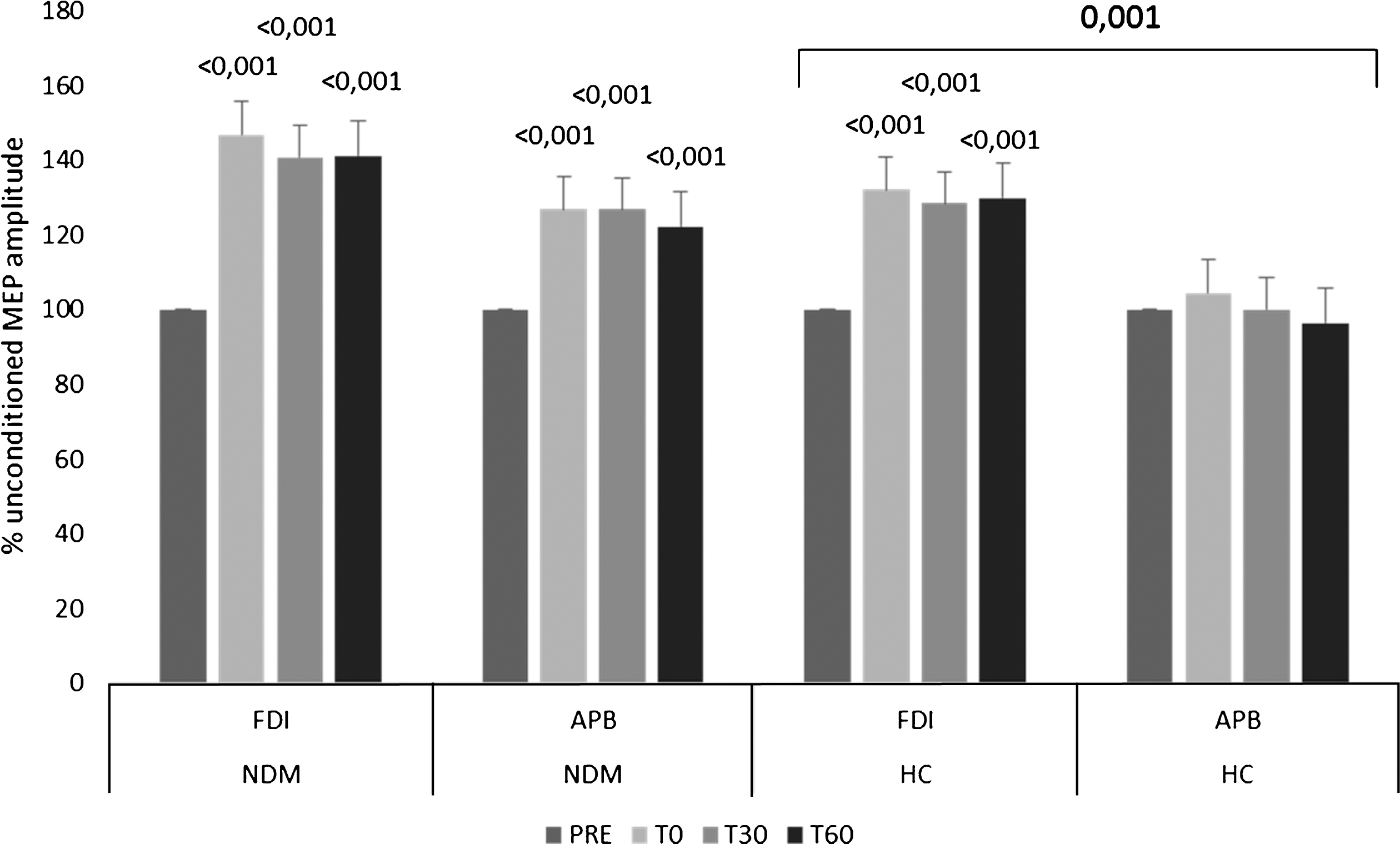
Repetitive paired associative stimulation aftereffects on MEP amplitude recorded from FDI and APB in patients with NDM and in HCs. Vertical error bars refer to SD. Superscript numbers refer to significant p-values at each interval as compared to baseline (p < 0.05), that upon square bracket to a significant muscle effect in HCs.
Then, we performed a cluster analysis to assess significant differences in terms of cortical excitability and synaptic plasticity depending on NDM type. As we did not observe relevant clinical, demographic, neurocognitive, neuropsychological, and electrophysiological differences between MC and PMC on-mexiletine (all interactions and effects, p > 0.05), we focused the cluster analysis on the electrophysiological differences with regard to mexiletine intake. There were no significant clinical-demographic differences between the subgroups with regard to both NDM type and mexiletine intake.
The on-treatment patients showed a minor impairment of topographically-specific rPAS aftereffects as compared to those off-treatment (time×muscle×mexiletine-intake F (3,99) = 5.1, p = 0.002; on-mexiletine time×muscle F (3,87) = 4.8, p = 0.003; off-mexiletine time×muscle F (3,27) = 5.4, p = 0.005). MEP increase got the highest values at T30 in the patients on mexiletine, whereas all the others showed the highest MEP amplitudes yet from T0 (Fig. 3). Finally, we found that MEP amplitude increase was not correlated with baseline MEP amplitude (Pearson’s correlation p = 0.1).
No correlations were found between
For the first ever time, our study demonstrates an involvement of cortical excitability and plasticity in patients with NDMs. In fact, the patients showed a greater increase in MEP amplitude at the I-O curve, a shorter CSP duration, and a loss of muscle surround inhibition as compared to the HCs, whereas both MEP amplitude at rest and RMT were not different from HCs values. These findings suggest a global cortical excitability increase in patients with NDMs, which was not related to both NDM type and myotonia severity. Further, patients with NDMs showed a greater MEP amplitude increase following rPAS and a loss of topographic specificity as compared to HCs, i.e., MEP amplitude increased in both muscles tested, which was not the case of HCs, who showed only a MEP amplitude increase from FDI muscle. Noteworthy, both magnitude and topographic specificity of rPAS aftereffects were independent from baseline MEP amplitude and the preservation or not of muscle surround inhibition. Altogether, these issues suggest that the cortical excitability abnormalities do not reflect a use-dependent plasticity (Liepert et al. 1998; Pascual-Leone et al. 1995; Shen et al. 2016; Shen et al. 2017). In other words, muscle hyperexcitability does not influence sensorimotor plasticity by means of, e.g., change in sensory feedback from muscle spindles (Oliveri et al. 1997; Bartel et al. 1984). Moreover, we did not find any correlation between neurophysiological parameters and the degree of myotonia. Therefore, the abnormalities of cortical excitability and sensorimotor plasticity may be considered as an additional epiphenomenon (i.e., a primary trait) of NDMs, rather than a consequence of muscle hyperexcitability, as formerly demonstrated in DMs (Oliveri et al. 1997; Portaro et al. 2017). The similarities in sensorimotor plasticity involvement between DMs and NDMs suggest that myotonia is part of a multiorgan impairment that may include the brain due to, at least in part, ion channel or kinase involvement. This issue is supported by the significant dependency of rPAS aftereffects by mexiletine intake. Mexiletine (1-(2,6-dimethylphenoxy)-2-aminopropane) is a class IB antiarrhythmic drug, commonly used to treat ventricular arrhythmias (Roden, 2001), long QT-3 syndrome (Schwartz et al. 1995), and neuropathic pain (Chabal et al. 1992), and it is also employed in neuroprotection (Taylor and Meldrum, 1995; De Luca et al. 1997a,1997b, 2000; Fenster and Comess, 1986; Cannon, 1996; Lehmann-Horn and Rüdel, 1996; Jackson et al. 1994; Wang et al. 2004). Moreover, mexiletine can reduce muscle fiber excitability, as suggested by in-vitro and in-vivo animal models (Desaphy et al. 2001; De Luca et al. 2004; Wang et al. 2004; Mohammadi et al. 2005). Therefore, it has also been used as a safe and effective anti-myotonia medication at 400 to 600 mg daily (Jackson et al. 1994; Lehmann-Horn and Jurkat-Rott, 1999; Logigian et al. 2010; Statland et al. 2012). The off-treatment patients (ten PMC) had poorer topographically specific rPAS aftereffects than the on-treatment ones (all MC and five PMC). The drug acts by blocking the fast Na+ channels (Apps, 1992), even at CNS level (Stys and Lesiuk, 1996). In particular, mexiletine inhibits the inward Na+ currents, thus reducing the rate of rise of the action potential. This blockage occurs by the binding of the drug to its high-affinity site on the a-subunit of the channel when opened or inactivated (Catterall, 1987; De Luca et al. 1991, 1997; Sunami et al. 1993). Moreover, mexiletine acts in a use-dependent modality, i.e., it occurs when cells are more active rather than in resting or tonic condition. This is likely due to changes in receptor affinity or in the access to the receptor, or both (Hille, 1977; Grant and Wendt, 1992; Catterall and Mackie, 2001). It is known that rPAS-induced synaptic plasticity depends on the level of Na+ channel activity related to NMDA-dependent long-term potentiation (LTP) mechanisms by means of spike-time dependent plasticity (Stefan et al. 2002; Carson and Kennedy, 2013). This depends, in turn, on a number of factors, including firing rate, number of coactive synaptic inputs, timing of the inputs, and postsynaptic voltage (Feldman, 2012). Given that the latter depends on Na+ influx (Lodish et al. 2000), which affects LTP and, thus, rPAS aftereffects, it is possible that mexiletine acts at post-synaptic level. Moreover, there is evidence that the generation and regulation of surround inhibition are mediated through GABAergic interneurons (Beck and Hallett, 2011). These also represent the main target of rPAS (Feldman, 2012), beyond cholinergic interneurons (Quartarone et al. 2006). These issues can explain the reason why mexiletine reverted the abnormal pattern of the surround inhibition. The patients with MC harbored a ClCN1 mutation. However, rPAS paradigm does not target Cl- channels, despite their putative role in regulating LTP mechanisms has been proposed (Farmer et al. 2013). Thus, the role of chloride channels concerning mexiletine effects deserves further studies. The main limitation of our study consists in the small sample of the off-treatment patients. Notwithstanding, NDMs are rare neuromuscular diseases with a very low prevalence (Horga et al. 2013). Therefore, also small sample studies could contribute to the current body of knowledge on NDMs. In conclusion, our findings suggest that patients with NDMs have an involvement of cortical sensory-motor plasticity that may be a trait of the disease. Such findings, combined to clinical, molecular, and functional characterization of the NMDs mutations (Portaro et al. 2015) may pave the way toward a better understanding of such diseases.
Authors’ declarations
Acknowledgments
N/A.
Conflict of interest
None of the authors has conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Funding
No funding to report.
Informed consent
Patients provided their written informed consent to study participation.