
Abstract
Stroke often leads to neuronal injury and neurological functional deficits. Whilst spontaneous neurogenesis and axon regeneration are induced by ischemic stroke, effective pharmacological treatments are also essential for the improvement of neuroplasticity and functional recovery after stroke. However, no pharmacological therapy has been demonstrated to be able to effectively improve the functional recovery after stroke. Bumetanide is a specific Na+-K+-Cl– co-transporter inhibitor which can maintain chloride homeostasis in neurons. Therefore, many studies have focused on this drug’s effect in stroke recovery in recent years. Here, we first review the function of Na+-K+-Cl– co-transporter in neurons, then how bumetanide’s role in reducing brain damage, promoting neuroplasticity, leading to functional recovery after stroke, is elucidated. Finally, we discuss current limitations of bumetanide’s efficiency and their potential solutions. These results may provide new avenues for further exploring mechanisms of post-stroke functional recovery as well as promising therapeutic targets for functional disability rehabilitation after ischemic stroke.
Introduction
Stroke remains the leading cause of morbidity and mortality for the aged person in the world. As the brain has a high plasticity potential, it can undergo self-repair and has the ability to compensate for functions lost after a stroke. These repair mechanisms include axonal and dendritic reorganization, altered excitability, neurogenesis and angiogenesis. However, these self-repair processes are not enough in order to reach satisfying recovery levels alone. To reduce the burden of neural injury and regain the neurological function, more strategies have been devised to enhance neuroplasticity after stroke, amongst which, some pharmacological treatments and rehabilitative therapies have been shown to be more promising to alter and improve behavioral recovery, even several days after the insult has occurred (Clarkson et al., 2011; Mu et al., 2017; Qu et al., 2015; Windle & Corbett, 2005; Zhao, Zhao, Xiao, Jolkkonen, & Zhao, 2013).
Na+-K+-Cl– co-transporters (NKCCs) are important neuronal function regulators, expressed in neurons throughout the brain, maintaining the homeostasis of chloride and neuronal excitatory potential (Cuomo et al., 2015). The major function of NKCCs is to regulate nerve injury and repair through GABAergic signaling. The loop diuretic bumetanide can specifically block the cation-chloride co-transporter-Na+-K+-Cl– cotransporter 1 (NKCC1) in the brain, and this function has been applied to the management of some nervous system disorders. Some clinical trials have demonstrated that low dose oral bumetanide may be effective in the treatment of Autism Spectrum Disorder in children (James, Gales, & Gales, 2019; Lemonnier & Ben Ari, 2010). It has also been shown that this drug can reduce seizure susceptibility as well as enhance the anti-seizure efficacy of phenobarbital in animal models (Brandt et al., 2018; Dzhala, Brumback, & Staley, 2008; Hu et al., 2017; Liu, Shangguan, Barks, & Silverstein, 2012; Wang et al., 2015), but evidence supporting its effectiveness in treating human epilepsy are still limited (Eftekhari et al., 2013; Kahle, Barnett, Sassower, & Staley, 2009; Pressler et al., 2015). A double-blind placebo-controlled longitudinal study has also demonstrated that bumetanide reduces refractory hallucinations, in schizophrenia patients, dramatically (Rahmanzadeh et al., 2017). Bumetanide seems to be a promising therapy for post-stroke recovery as well. Through NKCC1 inhibition, bumetanide protects the ischemic brain (Busse, Breder, Dinkel, Reymann, & Schröder, 2005; Luo et al., 2008; Pond, Galeffi, Ahrens, & Schwartz-Bloom, 2004), creating a favorable environment for neurogenesis (Xu et al., 2017; Xu et al., 2016) and axon growth (Mu et al., 2017), which may lead to improved outcomes after stroke. Here, we review the experimental studies of bumetanide treatment and use of this information as an example of how pharmacotherapy might promote recovery after stroke. We believe that this approach will be helpful in finding ways to translate the promising experimental evidence of this drug on stroke recovery into clinical practice in an efficient and safe manner.
The Na-K-Cl cotransporter (NKCC) and GABAergic signaling
The Na+-K+-Cl–-co-transporter (NKCC) is the main ion transporter in the central nervous system which aids the active transport of sodium, potassium, and chloride into cells. It has two isoforms encoded by two separate genes: one is NKCC1 (translated from the human gene, SLC12A1), which mediates the internal flow of chlorine ions; another one is NKCC2 (derived from the gene, SLC12A2), that mediates the outflow of chlorine ions (Isenring, Jacoby, Payne, & Forbush, 1998). NKCC1 exists in various types of tissues including brain, whilst NKCC2 is restricted, mostly to the kidney (Russell, 2000). Gamma-aminobutyric acid (GABA) is the major inhibitory neurotransmitter, related to the activation of chloride-permeable channels in the adult mammalian brain. The concentration of intracellular chloride determines the GABA mediated depolarization or hyperpolarization action of neurons (Cellot & Cherubini, 2013). Therefore, the different NKCC expression patterns on cell membrane between mature and immature neurons may cause the diverse actions of GABAergic signaling (Fig. 1). Where the expression of NKCC2 on the cell membrane of mature neurons is higher than that of NKCC1, the concentration of intracellular Cl– is lower than that of extracellular Cl– (Rivera et al., 1999). Therefore, it causes a GABA mediated cell membrane hyperpolarization that ultimately causes synaptic inhibition (Pfeffer et al., 2009). In contrast, in neural precursor cells and immature neurons, when the expression of NKCC1 on the cell membrane is higher than that of NKCC2 (Ben-Ari, Gaiarsa, Tyzio, & Khazipov, 2007; Young, Taylor, & Bordey, 2011), the NKCC1-mediated Cl- accumulation causes GABAergic excitation, cell membrane depolarization that finally leads to synaptic excitation (Pfeffer et al., 2009; Yamada et al., 2004).
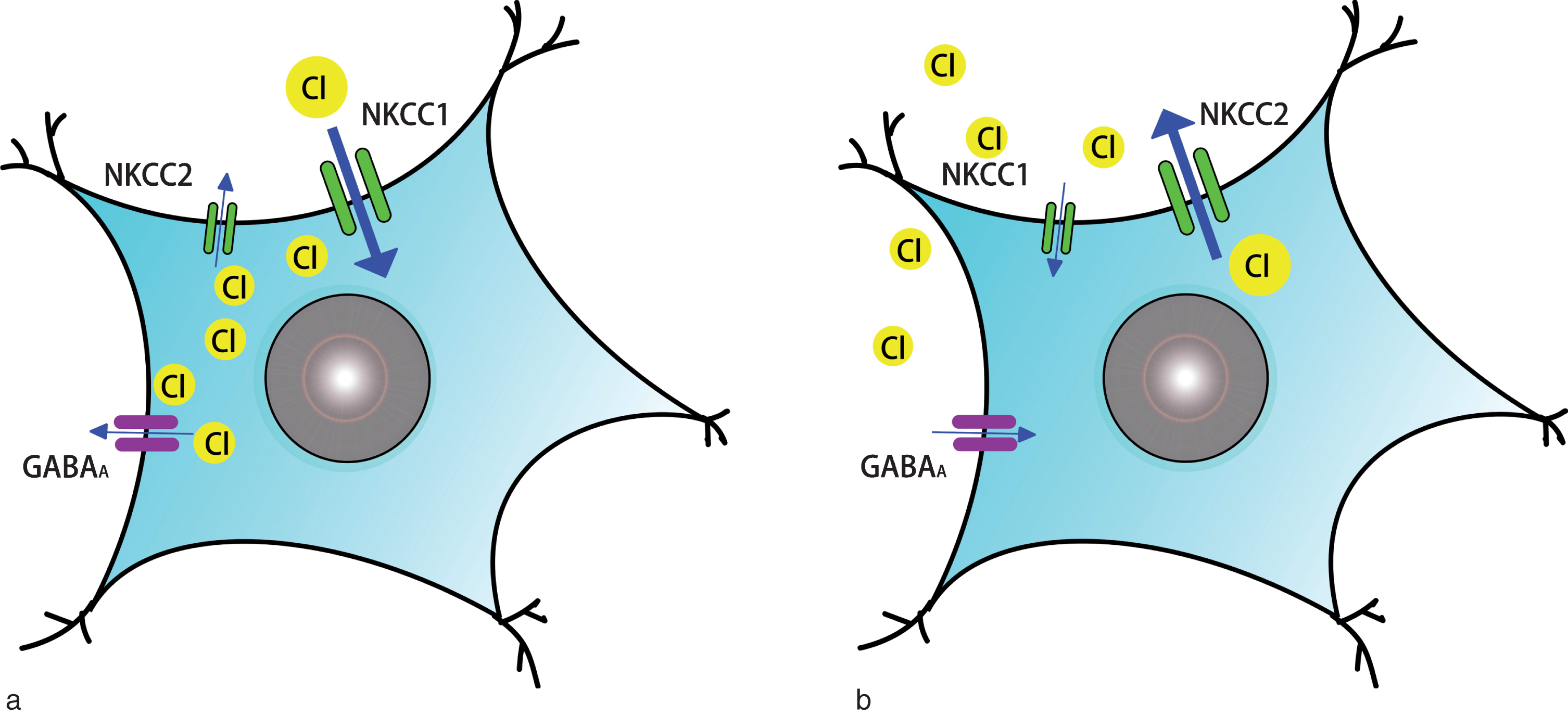
Different Na+-K+-Cl– co-transporter expression patterns and actions of GABAergic signaling between immature (or ischemic) neurons and mature neurons. A. NKCC expression pattern and action of GABAergic signaling on immature or ischemic neurons: the expression of NKCC1 on cell membrane is higher than that of NKCC2, NKCC1-mediated intracellular Cl– accumulation causes GABAergic excitation. B. NKCC expression pattern and action of GABAergic signaling on mature neurons: the expression of NKCC2 on cell membrane is higher than that of NKCC1, the intracellular Cl– concentration is lower than that of extracellular Cl–, which causes GABAergic inhibition.
The expression of NKCC can also be altered under some specific situations, such as cerebral ischemia. Studies have demonstrated that an increased expression of NKCC1 and decreased expression of NKCC2 in neurons after stroke, which is similar to that found in immature neurons (Jaenisch, Witte, & Frahm, 2010; Wang, Huang, He, Ruan, & Huang, 2014; Yan, Dempsey, Flemmer, Forbush, & Sun, 2003). Arginine vasopressin (AVP), hypoxia, p38 and JNK MAP kinases are factors involved in the activation and upregulation of NKCC1 in the ischemic brain (Foroutan, Brillault, Forbush, & O’Donnell, 2005; O’Donnell, Lam, Tran, Foroutan, & Anderson, 2006; O’Donnell, Duong, Suvatne, Foroutan, & Johnson, 2005; Wallace, Jelks, & O’ Donnell, 2012). This change in NKCC expression occurs after ischemic stroke and may be responsible for the increased level of Na+ and Cl– in neurons and thus GABA-mediated depolarization, thereby contributing to the neuronal hyperexcitability and cell swelling observed after cerebral ischemia (Jaenisch, Witte, & Frahm, 2010; Yan, Dempsey, & Sun, 2001). NKCC1 is also involved in ischemic neuron damage by disrupting endoplasmic reticulum Ca2 + homeostasis (Chen et al., 2008). Therefore, NKCC1 is an important contributor to cerebral edema formation and neuron damage in ischemic stroke. Through docking to the binding site at the trans-membrane region of NKCC1, bumetanide blocks the activity of this ion channel and thus decreasing internal chloride concentrations in neurons (Jaggi, Kaur, Bali, & Singh, 2015). As a result, bumetanide may protect these cells and serve as a potential method for stroke recovery by blocking NKCC1 activity on neurons.
Bumetanide reduces edema formation and brain damage after stroke
Brain edema, an important complication of ischemic stroke, is defined as an increase in brain tissue volume caused by an increase in its fluid content (Katzman et al., 1977). It has been reported that intravenous administration of bumetanide can inhibit Na+-K+-Cl– cotransporter action on both luminal membrane of the blood-brain barrier and neurons, reducing edema formation by up to 70% in a middle cerebral artery occlusion (MCAO) rat model (O’Donnell, Tran, Lam, Liu, & Anderson, 2004; Yan, Dempsey, & Sun, 2001). Bumetanide has more inhibition on edema formation in younger mice and rats with higher levels of estrogen, which shows that this drug’s effects is influenced by these factors (Liu, Akella, Benashski, Xu, & McCullough, 2010; O’Donnell, Lam, Tran, Foroutan, & Anderson, 2006). Furthermore, bumetanide treatment even can reduce edema exacerbated by hyperglycemia, brain Na+ uptake and ischemic injury in a rat MCAO model (Yuen et al., 2018). Aquaporins (AQP) are the most abundant water channel in brain which may alter neurological outcomes through the regulation of edema, astrocyte and glial scar formation as well as the modulation of inflammation and signaling functions (Vella, Zammit, Di Giovanni, Muscat, & Valentino, 2015). Some studies have also demonstrated that bumetanide may ameliorate stroke-related cerebral edema formation and improve neurological outcomes through AQP4 and the perivascular pool of AQP4 (King & Agre, 1996; Migliati et al., 2009; Migliati et al., 2010).
Neuronal damage is another common complication after stroke, which often causes the irreversible loss of neuronal system function. Bumetanide with its ability to maintain ionic homeostasis, through NKCC1 inhibition, attenuates neuronal damage during both ischemia and ischemia reperfusion conditions (Chen, Luo, Kintner, Shull, & Sun, 2005; Luo et al., 2008; Wang, Huang, He, Ruan, & Huang, 2014). In addition, the size of cerebral infarction can be reduced by this drug when applied before or during the acute period of stroke, rather than in the convalescent period (Mu et al., 2017). In addition, bumetanide can also protect white matter from ischemic damage. After genetic ablation of NKCC1 in mice (an ortholog of human NKCC1), reduced damage to both grey and white matter was found in a model of focal cerebral ischemia (Chen, Luo, Kintner, Shull, & Sun, 2005). Moreover, the white matter lesions, induced by chronic cerebral hypoperfusion, can be reduced significantly by bumetanide action through enhancing progenitor cells proliferation, a function that might be mediated by the regulation of the MAPK signaling dependent pathways (Yu et al., 2018). Bumetanide also may exert a protective effect on white matter through preventing periventricular leukomalacia in developing brains and prevents neurons from degeneration after hypoxia ischemic injury (Jantzie et al., 2015). Thus, we speculate that invoking NKCC1 inhibition after ischemia in vivo, we might also exert a protective effect with similar mechanisms.
In in vitro ischemia models of oxygen-glucose deprivation and glutamate induced excitotoxicity, bumetanide has a neuronal protective function, that is latent only in the initial stage of neurotoxicity (Beck, Lenart, Kintner, & Sun, 2003). The protective effect of NKCC1 inhibition following oxygen-glucose deprivation is mediated by the recovery of cellular energy stores and the promotion of hippocampal cell survival (Pond, Galeffi, Ahrens, & Schwartz-Bloom, 2004). Therefore, we speculate that reducing NKCC1 activity, following ischemic stroke, might also show a neuroprotective effect through similar mechanisms in vivo. It has been shown that, after traumatic brain injury, NKCC1 activation leads to p75NTR upregulation, which renders neurons dependent on BDNF for survival and resistant to apoptosis (Shulga et al., 2008). Bumetanide may upregulate the neural protective NKCC2 which leads to BDNF independent survival by blocking the up-regulation of pan-neurotrophin receptor p75NTR in the post-traumatic brain injury, thus rescuing injured neurons that leads to a better behavioral recovery (Alder et al., 2015; Shulga et al., 2012). p75NTR is also induced in both in vivo and in vitro ischemic models and causes neuronal apoptosis following ischemia (Irmady et al., 2014). It is therefore plausible that this drug might show a similar protective role, by the same mechanisms, in ischemic stroke induced neuron injury in vivo, but the exact details need to be explored and proven by future studies.
Post-stroke bumetanide administration promotes neurogenesis
It has been demonstrated the ischemic stroke can robustly increase adult neurogenesis, in both the SVZ and SGZ, by stimulating neural precursor cell (NPCs) proliferation during 4 days to 2 weeks after stroke (Arvidsson, Collin, Kirik, Kokaia, & Lindvall, 2002; Doetsch, García-Verdugo, & Alvarez-Buylla, 1997; Jin et al., 2001; Morshead et al., 1994; Yamashita et al., 2006). The four essential steps of neurogenesis after stroke are proliferation, migration, differentiation, followed by survival or integration (Christie & Turnley, 2012). After focal proliferation, the stroke-induced neuroblasts migrate towards the injured site, where they differentiate into mature neurons, establish appropriate synapse with other neurons and finally integrate into the neuronal circuitry (Carmichael, Kathirvelu, Schweppe, & Nie, 2017; Jiang, Gu, Brännström, Rosqvist, & Wester, 2001; Parent, Vexler, Gong, Derugin, & Ferriero, 2010). Although ischemia-induced neurogenesis may compensate for the loss of functional neurons and contribute to stroke recovery, normally it is limited in success in recovering function. One of the reasons behind this is the low survival rate of high-quality newborn neurons. It has been observed that 80% or more of the new cells died during this time interval (Arvidsson, Collin, Kirik, Kokaia, & Lindvall, 2002; Thored et al., 2006), and only about 0.2% of the neurons killed during the ischemic event are replaced by the new ones at 6 weeks after insult (Arvidsson, Collin, Kirik, Kokaia, & Lindvall, 2002). Another limitation is that neurons generated from SVZ possibly cannot replace the broad spectrum of the subtypes of neurons damaged or lost by stroke (Inta & Gass, 2015). Therefore the insufficiency of spontaneous neurogenesis leads us to explore pharmacological methods in order to compensate for the lost brain function induced by ischemic stroke.
There is evidence indicating that bumetanide participates in several critical steps of post-stroke neuronal regeneration. Recent studies have demonstrated that post-stroke treatment with bumetanide during chronic phase significantly enhances the production of neuroblasts, both in the SVZ and the DG of the hippocampus, improves dendritic development of neural precursor cells, creates a favorable environment for the long-term survival of newborn neurons in the peri-infarct area, as well as enhancing the migration of neuroblasts towards the infarct area (Xu et al., 2017; Xu et al., 2016). Furthermore, bumetanide administration can also significantly enhance cell proliferation and dendritic development of newborn dentate gyrus cells after neonatal seizures, caused by hypoxic-ischemic encephalopathy (Wang et al., 2015). However, it did not alter the neurogenesis in normal rats, indicating it has different effects under physiological and pathological conditions (Xu et al., 2017). So far, the underlying mechanism of these effects of bumetanide on neurogenesis is unclear and needs further study.
Post-stroke bumetanide administration promotes axonal growth and possible synaptic remodeling
Axonal growth and synaptic remodeling after stroke are important steps in recovery from this affliction. Promoting functional and anatomical axonal reorganization can improve the recovery outcomes after stroke (Benowitz & Carmichael, 2010). Both axonal sprouting from the peri-infarct region to connected cortical areas and axonal sprouting from the contralateral cortex to the de-afferented side of the cervical spinal cord are associated with post-stroke recovery (Bachmann, Lindau, Felder, & Schwab, 2014; Overman et al., 2012; Wahl et al., 2014). In a murine model of sensorimotor cortex injury, better performance in grid walking and cylinder tests is correlated with GABAAR downregulation and axon sprouting from the contralesional cortex into the denervated side of the cervical spinal cord, whilst GABAAR agonist treatment significantly inhibited axonal sprouting, resulting in worse recovery outcomes (Lee, Ueno, & Yamashita, 2011). The NKCC1 inhibitor bumetanide can regulate the intracellular Cl- level thus causing a reversion of the GABAA mediated depolarization and maintain neuronal chloride homeostasis after injury (Dzhala et al., 2005), which may possibly accelerate axonal sprouting after stroke. The extent of axonal sprouting is also limited by the unfavorable local microenvironment, with intrinsic growth-inhibitory molecules like Nogo-A (Pernet & Schwab, 2012) or the lack of support from appropriate neurotrophic agents. After bumetanide treatment, the level of NKCC1 and growth-inhibitory Nogo-A is decreased, accompanied with an increased level of KCC2 and BDNF, as well as enhanced synaptic plasticity, creating a favorable and protective environment for neuronal growth, causing an increase in fiber sprouting into the denervated cervical spinal cord (Mu et al., 2017). It has also been shown that chronic bumetanide treatment increases the expression of axonal growth factors and reactive synaptogenesis markers, PSD-95 and vGlut-1, causing enhanced functional corticospinal tract fibers crossing the midline of spinal cord (Mu et al., 2017), which finally lead to synaptic remodeling after cerebral ischemia. OPCs play an essential role in myelination and also provide a viable substrate for the damaged axons to regenerate in local microenvironments (Lee et al., 2012; Wilkins, Majed, Layfield, Compston, & Chandran, 2003), which may finally result in a better performance in motor learning and cognitive function (McKenzie et al., 2014; Xiao et al., 2016). Bumetanide may also promote remyelination after cerebral ischemic by the regulation of OPCs activity: After oxygen and glucose deprivation, the proliferative ability of OPCs is decreased and a cell cycle progression occurs, bumetanide-induced NKCC1 inhibition attenuates the downregulation in proliferative activity and cell cycle progression arrest of OPCs after cerebral ischemia through p-38 MAPKs (Fu et al., 2015).
Bumetanide enhances functional recovery after stroke
It has been established that NKCC1 inhibition is beneficial for the recovery after many neurological disorders, including stroke. Suppressing NKCC1 activity with bumetanide has been demonstrated to play a neuronal protective role and improve neurological outcomes after traumatic brain injury (Shulga et al., 2012; Zhang et al., 2017). Bumetanide, as an inhibitor of NKCC1, has effects on many central nerve system disorders. It has been shown that bumetanide can reverse alterations in sociability and functional brain connectivity caused by early-life seizures (Holmes et al., 2015), and restore Cl– currents (ECl), synaptic plasticity and hippocampus-dependent memory in adult Down syndrome model mice (Deidda et al., 2015). Coadministration of phenobarbital and bumetanide augments the neuroprotective efficacy of the former drug and hypothermia in a neonatal hypoxia-ischemia rat model (Liu, Shangguan, Barks, & Silverstein, 2012). Until now, whether bumetanide can alter the behavioral recovery in stroke has been uncertain. A study focusing on GABA signaling show that pharmacologically or genetically lowering the α5 or δ-subunit-containing GABAARs or sustained low-dose GABAA antagonism treatment in mice lead to better performance in the grid walking and cylinder tasks, which suggests that reducing excessive GABA-mediated tonic is beneficial for post-stroke functional recovery (Clarkson, Huang, Macisaac, Mody, & Carmichael, 2010). WNK3 (with no lysine) kinase is an upstream regulator of NKCC1, which can activate NKCC1 through phosphorylation (Begum et al., 2015; Kahle et al., 2005). Recent studies have indicated a decrease in NKCC1 stimulatory sites, as well as a decrease in NKCC1 expression on cell surface induced by WNK3 knockdown could reduce neural damage and accelerate behavioral recovery (Begum et al., 2015; Bhuiyan et al., 2017). As mentioned above, bumetanide protects brain from ischemic injury through NKCC1-mediated GABAergic signaling in the acute phase (O’Donnell, Tran, Lam, Liu, & Anderson, 2004; Chen, Luo, Kintner, Shull, & Sun, 2005). So treatment with bumetanide, which has a similar effect of GABAA receptor antagonist and WNK3 knockout, may promote functional recovery after stroke via neuroprotection. Neuroplasticity including neurogenesis and axonal sprouting, is an important contributor to stroke recovery in the chronic phase (Hermann & Chopp, 2012). There are emerging evidences that demonstrated the behavioral effects of bumetanide correlated with neural plasticity in rodent models: Bumetanide treatment starting from 7 days after stroke enhances neurogenesis and axon sprouting and improves the sensorimotor recovery in stroke rats (Mu et al., 2017; Xu et al., 2017). In a model of focal ischemia, rats in a group where bumetanide was administered showed better learning and memory functions as assessed by escape latency and platform crossing in a water maze test (Xu et al., 2016). All these studies suggest that beneficial effects of bumetanide for stroke recovery have been indicated possibly through promoting neuroprotection during the acute phase and stimulating neural plasticity during the chronic phase.
Limitations of bumetanide treatment and their potential solutions
When being applied to treat neurological diseases, bumetanide has shown some disadvantages that may limit its application to some extent. Firstly, the highly diuretic effect of bumetanide might lead to potassium loss and cause hypokalemic alkalosis (Greenberg, 2000). Secondly, as a result of its benzoic acid moiety, bumetanide is highly ionized at physiological pH, highly bound to plasma proteins, which may limit its penetration into the brain and cause its accumulation in the kidney (Brandt, Nozadze, Heuchert, Rattka, & Löscher, 2010; Cohen et al., 1976; Kim & Lee, 2001; Tollner et al., 2014). Furthermore, pharmacokinetic analysis indicates that bumetanide is eliminated in adult rats extremely rapidly, causing the levels of the drug to fall below those needed to inhibit NKCC1 in the brain (Kim & Lee, 2001). Organic anion transporter 3 (Oat3), organic anion-transporting polypeptide (Oatp) Oatp1a4 and multidrug resistance protein 4 may cause restricted passive diffusion and active efflux transport, which finally lead to the extremely low brain concentrations, after systemic administration of this drug (Römermann et al., 2017).
Although after systemic administration, brain concentrations and the brain to plasma ratio of this drug are extremely low, bumetanide is still effective for many central nervous system disorders, including ischemic stroke. One possible explanation for this is bumetanide may target tissues outside blood-brain barrier or other targets in central nervous system rather than NKCC1 (Wang et al., 2015). Some existing studies may support this explanation: NKCC1 was highly expressed on the luminal side of the BBB and was upregulated after ischemic stroke (Kahle, Simard, Nahed, Jones, & Sun, 2009; O’Donnell, Tran, Lam, Liu, & Anderson, 2004). As a result, systemically administrated bumetanide can directly inhibit NKCC1, reduce edema formation and lead to better functional recovery without passing the BBB (O’Donnell, Tran, Lam, Liu, & Anderson, 2004). Another possible explanation may be that the blood-brain barrier in the SVZ and circumventricular organs (CVO) niches is leakier than elsewhere in the brain and this can be further elevated after stroke (Lin et al., 2018). The more permeable BBB in these niches may allow neural stem cells to get more access to systemic factors such as systemic administrated bumetanide, so the low brain concentration may not limit the neurogenesis process following bumetanide treatment.
More penetrable lipophilic and uncharged prodrugs that can be converted into bumetanide have been designed in order to solve the low penetration of this drug into the CNS: In chronic epilepsy models of mice and rats, an ester prodrug N,N-dimethylaminoethylester (BUM5) treatment results in higher levels of bumetanide in the brain and displays a more effective anti-seizure function (Erker et al., 2016; Tollner et al., 2014). Probenecid inhibits efflux transporter organic anion transporter 3 (oat3) both in vivo and in vitro, thus may also serve as a potential solution for this low BBB passage of bumetanide by inhibiting its elimination (Donovan, O’Brien, Boylan, Cryan, & Griffin, 2015; Donovan, Schellekens, Boylan, Cryan, & Griffin, 2016). However bumetanide concentrations in brain and the brain to plasma ratio are not upregulated to a great extent, following administration of these existing bumetanide prodrugs and probenecid treatment (Donovan, O’Brien, Boylan, Cryan, & Griffin, 2015; Erker et al., 2016; Tollner et al., 2014), which may limit their application. A novel NKCC1 Inhibitor STS66, which is a prodrug of bumetanide, can penetrate BBB more easily, and appears to be more efficient in reducing ischemic infarction, swelling, and neurological deficits in the model of MCAO (Huang et al., 2019). Several new research directions are suggested by the current state of our knowledge. First, the target of bumetanide in stroke therapy need to be clarified. Second, whether the level of bumetanide in brain can be elevated and whether the neural plasticity and functional recovery can be promoted after administration of bumetanide prodrugs or probenecid remain to be investigated. Finally, further exploration of this topic should focus on the development of more permeable NKCC1 inhibitors for stroke recovery in the future.
Conclusion
After ischemic stroke, chloride homeostasis is disrupted by the upregulated expression of NKCC1 and post-stroke edema is formed via the perivascular pool of the water channel AQP. Bumetanide specifically blocks NKCC1 and AQP, leading to a reduction in edema size and neuronal injury. Moreover, bumetanide administration may create a favorable microenvironment for neurogenesis, axon regeneration and synaptic restoration, which contribute to the improvement of behavioral and functional recovery. The exact mechanisms for these beneficial effects of bumetanide needs to be fully explored in further studies, which will provide a new therapeutic target aimed at reducing the neural damage that occurs after stroke.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work is supported by The Liaoning Province Key Research and Development Project Critical Project (No. 2017225005, Chuansheng Zhao); The Shenyang Municipal Bureau of Science and Technology International Exchange and Cooperation Project (No. 17-129-6-00, Chuansheng Zhao); China Medical University High-level Innovation Team Training Plan (No. 2017CXTD02, Chuansheng Zhao).