
Abstract
Background:
Permanent loss of vital functions after central nervous system (CNS) injury occurs in part because axons in the adult mammalian CNS do not regenerate after injury. PTEN was identified as a prominent intrinsic inhibitor of CNS axon regeneration about 10 years ago. The PTEN negatively regulated PI3K-AKT-mTOR pathway, which has been intensively explored in diverse models of axon injury and diseases and its mechanism for axon regeneration is becoming clearer.
Objective:
It is timely to summarize current knowledge about the PTEN/AKT/mTOR pathway and discuss future directions of translational regenerative research for neural injury and neurodegenerative diseases.
Methods:
Using mouse optic nerve crush as an in vivo retinal ganglion cell axon injury model, we have conducted an extensive molecular dissection of the PI3K-AKT-mTORC1/mTORC2 pathway to illuminate the cross-regulating mechanisms in axon regeneration.
Results:
AKT is the nodal point that coordinates both positive (PI3K-PDK1-pAKT-T308) and negative (PI3K-mTORC2-pAKT-S473) signals to regulate adult CNS axon regeneration through two parallel pathways, activating mTORC1 and inhibiting GSK3β. However, mTORC1/S6K1-mediated feedback inhibition after PTEN deletion prevents potent AKT activation.
Conclusions:
A key permissive signal from an unidentified AKT-independent pathway is required for stimulating the neuron-intrinsic growth machinery. Future studies into this complex neuron-intrinsic balancing mechanism involving necessary and permissive signals for axon regeneration is likely to lead to safe and effective regenerative strategies for CNS repair.
Introduction
CNS axon injury and degeneration is a common feature of trauma and neurodegenerative diseases, which often cause life-long neurological deficits because CNS axons fail to regenerate in adult mammals (Fitch & Silver, 2008; Goldberg, Klassen, Hua, & Barres, 2002; Schwab & Bartholdi, 1996). Extensive evidence indicates that the intrinsic regenerative capacity of CNS neurons is diminished after development; reactivation is critically important for CNS axon regeneration (Park, Liu, Hu, Kanter, & He, 2010) (Benowitz, He, & Goldberg, 2017; Mahar & Cavalli, 2018). Mouse retinal ganglion cell (RGC) and optic nerve (ON) is a simple but robust in vivo system to study neural injury and diseases. Combining ON crush injury with adeno-associated virus (AAV)-mediated RGC gene targeting, we identified phosphatase and tensin homolog (PTEN) as an important regulator of the intrinsic regenerative signaling of RGCs (Park et al., 2008). Many additional signal transduction pathways and transcriptional factors have since been linked with CNS axon regeneration (Benowitz et al., 2017; Mahar & Cavalli, 2018; Zhang, Yang, Huang, Sun, & Hu, 2018). We and others have conducted an extensive molecular dissection of the PTEN related signal transduction pathways, which revealed the roles of cross-regulating mechanisms of PTEN/PI3K/mTORCs/AKT/GSK3β and their downstream effectors in ON regeneration (Al-Ali et al., 2017; Guo, Snider, & Chen, 2016; Huang et al., 2019; Miao et al., 2016; Park et al., 2008; L. Yang et al., 2014).
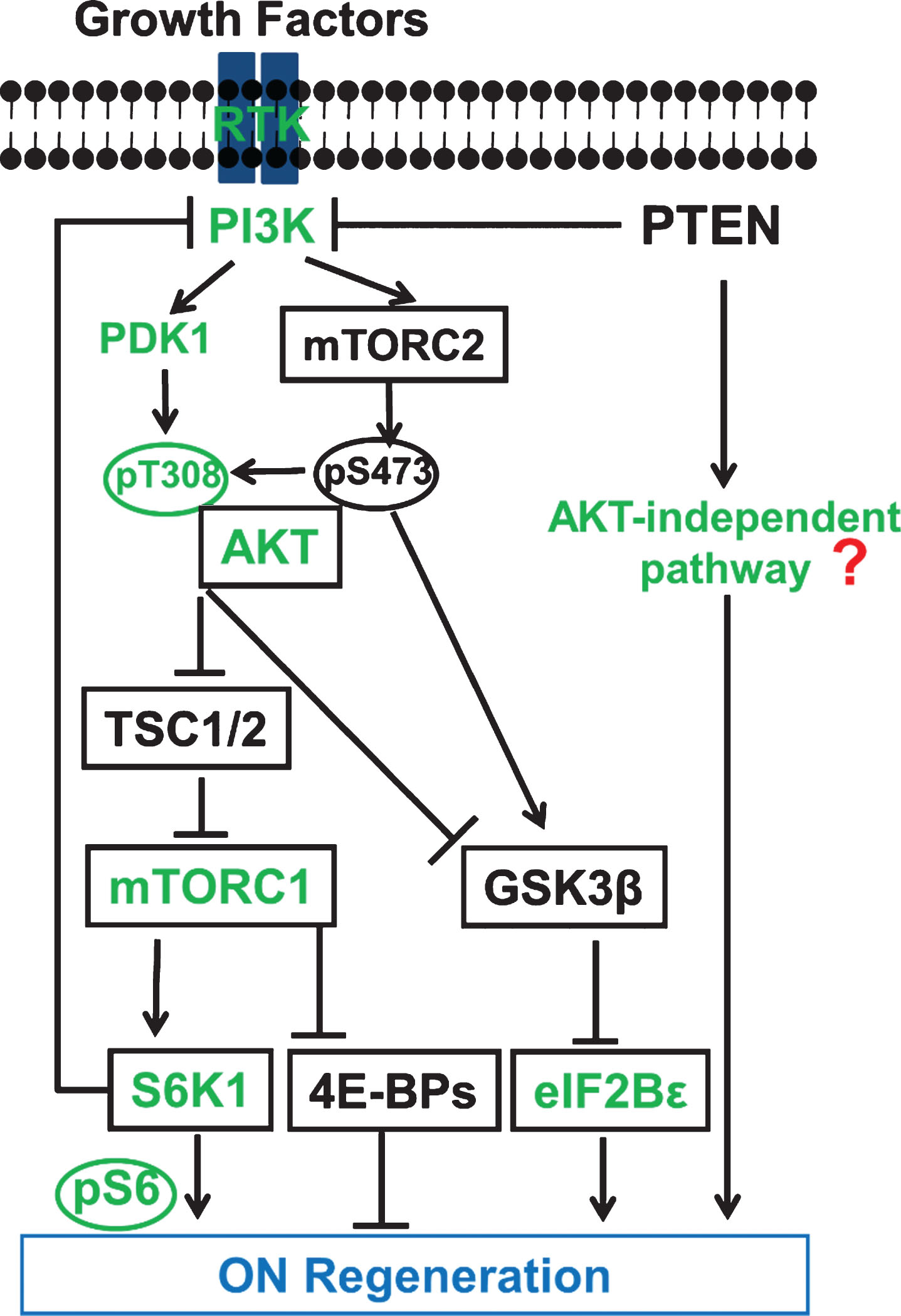
Schematic illustration of the PTEN regulated PI3K-AKT-mTOR signaling pathways in CNS axon regeneration. AKT is positively regulated by the PI3K/PDK1 pathway through phosphorylation of T308, and negatively regulated by the PI3K/mTORC2 pathway through phosphorylation of S473; its subsequent actions include at least partial regulation of GSK3β phosphorylation/inhibition. Both AKT downstream effectors, activation of mTORC1 and phosphorylation/inhibition of GSK3β, synergistically promote axon regeneration; inhibition of GSK3β alone is also sufficient for axon regeneration, at least partially through eIF2Bɛ. The activation of mTORC1 and its substrates 4E-BP and S6K is necessary but not sufficient for potent axon regeneration. Feedback inhibition from mTORC1 and S6K1 keeps AKT activation after PTEN deletion to a minimum. The question mark represents unknown effectors downstream of PTEN that are AKT-independent and sufficient to initiate CNS axon regeneration, and which potentially interact with the translational targets of mTORC1 to promote potent axon regeneration. Green color-coated molecules are pro-axon regeneration and black color-coated molecules are anti-axon regeneration.
Proper translational control is crucial for normal cell growth. Not surprisingly, multiple levels of crosstalk and feedback regulation among these important signaling molecules tightly regulate this master growth control pathway. One of the key feedback inhibitory mechanisms is that mTORC1 keeps AKT and mTORC2 activities in check by inhibiting RTK/PI3K directly or through its effector S6K1 and insulin receptor substrate 1 (IRS-1) (Laplante & Sabatini, 2012; Rozengurt, Soares, & Sinnet-Smith, 2014). mTORC2 may also counteract mTORC1: its defining component, RICTOR, inhibits mTORC1 (Chen et al., 2010), as does its downstream effector, AKT-pS473, at least in β-cells (Gu, Lindner, Kumar, Yuan, & Magnuson, 2011). AKT appears to be the nodal point that acts as the key substrate of both PI3K-PDK1 and mTORC2, and also as the critical upstream regulator of mTORC1 and glycogen synthase kinase 3 (GSK-3β) to control cell survival, growth, and proliferation (Manning & Cantley, 2007). AKT also mediates neuronal survival through transcription control (Brunet, Datta, & Greenberg, 2001). Inhibition of pro-apoptotic transcription factors, FOXOs and p53, or activation of anti-apoptotic CREB and NF-κB, by AKT protects neurons (Brunet et al., 2001; Read & Gorman, 2009).
RGCs relay visual information from retina to brain through ON, which is formed by the unidirectional RGC projection axons. Intravitreal AAV injection expresses transgenes specifically and efficiently in adult RGCs, in a spatially and temporally controlled manner can overcome developmental issues associated with germ line manipulation (Hu, 2015; Zhang et al., 2018). Our initial efforts to understand the intrinsic mechanisms of regenerative failure led us to postulate that CNS neurons tightly regulate the evolutionarily conserved molecular pathways that control cell growth to prevent overgrowth when development is complete. The initial effort to screen several evolutionarily conserved molecular pathways that control cell growth (Weinberg, 2007) using the mouse ON crush model revealed that deletion of PTEN, but not Rb (retinoblastoma), P53, Smad4, or LKB1 (liver kinase B1), promotes significant ON regeneration (Park et al., 2008). Similar axon regeneration phenotypes after PTEN inhibition were subsequently reported in mouse cortical motor neurons (Jin et al., 2015; Liu et al., 2010), drosophila sensory neurons (Y. Song et al., 2012) and C. elegans motor neurons (Byrne et al., 2014), indicating the critical role of PTEN in axon regeneration. However, because PTEN is a tumor suppressor gene, uncontrolled deactivation of PTEN and activation of mTOR and protein synthesis may produce severe negative consequences, such as tumor formation and cognitive impairment (Zoncu, Efeyan, & Sabatini, 2011). Since not all tumor suppressor genes are involved in axon regeneration (Park et al., 2008), the unique role of PTEN in determining the neuronal intrinsic regenerative ability motivated us to carry out an extensive molecular dissection of PTEN signaling. Our goal was to comprehensively understand the roles of its downstream signaling molecules in ON regeneration and to differentiate these roles from the contribution of PTEN signaling to tumorigenesis.
mTORC1 plays a necessary but not sufficient role in ON regeneration
The observation that PTEN deletion constitutively activates mTORC1 and that rapamycin, an inhibitor of mTORC1, blocks PTEN knockout (KO)-induced ON regeneration (Park et al., 2008) indicates that mTORC1 activation is required for axon regeneration. Our studies also show that blocking mTORC1 activity by deletion of a key mTORC1 component, RAPTOR (regulatory associated protein of mTOR) (Guertin et al., 2006; Laplante & Sabatini, 2012), in RGCs, significantly decreases PTEN KO-induced ON regeneration (Huang et al., 2019). Additional support for the necessary role of mTORC1 in ON regeneration comes from our studies with its downstream effectors: a 4E-BP1 mutant (4E-BP1-4A) and a S6K1 mutant (S6K1-T229A) that cannot be phosphorylated by mTORC1 largely block PTEN KO-induced ON regeneration (L. Yang et al., 2014). On the other hand, we found that over-expression of the constitutively active mutant of S6K1 significantly increases RGC cell size after ON crush but only modestly promotes axon regeneration; and that double deletion of 4E-BP1 and 4E-BP2 in RGCs does not promote axon regeneration, indicating the necessary but insufficient role of mTORC1 downstream effectors in axon regeneration (L. Yang et al., 2014). We also observed a similar outcome with TSC deletion: mTORC1 is activated to a greater extent than by PTEN deletion, but there is very little axon regeneration (Park et al., 2008). These results lead us to conclude that mTORC1 activation itself is necessary for axon regeneration but has only minimal effect on initiating axon regeneration. The possibility that other substrates of mTORC1 in addition to S6K1 and 4E-BP may be sufficient to induce significant axon regeneration cannot be ruled out. However, we think it more likely that the mTORC1 pathway essentially plays a necessary role in axon regeneration and that a key permissive signal downstream of PTEN is required to trigger the neuron-intrinsic growth machinery.
AKT coordinates positive signals from PI3K-PDK1 and negative signals from mTORC2 in regulating mTORC1 activation and GSK3β phosphorylation for ON regeneration
AKT is downstream of PTEN/PI3K but upstream of mTORC1 (Fig. 1). AKT1 and AKT2 are expressed in almost all tissues, but AKT3 is the predominant isoform in the brain (Easton et al., 2005). The report that deletion only of AKT3 reduces brain size (Easton et al., 2005) indicates a specific role in CNS growth control. We quantitated the expression levels of the three AKT isoforms in RGCs and identified their distinct roles in axon regeneration. The results provide strong evidence of the unique properties of AKT3 in retina: AKT1 and AKT3 are the major isoforms of AKTs in RGCs, but activation of AKT3 promotes significantly greater RGC survival and ON regeneration than activation of AKT1 (Miao et al., 2016). We suspect RGC molecules that are regulated by AKT3 but not AKT1 may contribute to this difference in axon regeneration. Identification of AKT3-specific substrates in neurons is actively pursued in the field. We confirmed that phosphorylation of AKT-T308 and the kinase activity of AKT are essential for mTORC1 activation and axon regeneration by overexpressing the kinase dead mutant or the T308A mutant of AKT in RGCs. In contrast, AKT-S473A mutant produces even more robust axon regeneration than wildtype AKT, indicating that AKT-S473 phosphorylation exerts a negative influence on axon regeneration (Miao et al., 2016). This surprising finding of opposite roles in axon regeneration implies that pT308 and pS473 of AKT act on different substrates or regulate the same substrates differentially. Phosphorylation of GSK3β-S9 by AKT inhibits GSK3β activity, which is critical for neuronal polarization, axon branching, and axon growth (Kim, Hur, Snider, & Zhou, 2011). The finding that pGSK3β-S9 is significantly increased after blocking mTORC2 or overexpressing AKT3-S472A mutant suggests that GSK3β is one of the AKT effectors that are differentially regulated by pAKT-T308 and pAKT-S473 (Miao et al., 2016). The results of our studies using GSK3β-S9A mutant and GSK3β KO mice provide additional evidence that GSK3β inhibits axon regeneration (Miao et al., 2016) and thus definitively resolve the contradictory reports in the literature regarding its role in CNS axon regeneration (Abe, Borson, Gambello, Wang, & Cavalli, 2010; Christie, Webber, Martinez, Singh, & Zochodne, 2010; Dill, Wang, Zhou, & Li, 2008; Gobrecht, Leibinger, Andreadaki, & Fischer, 2014; Saijilafu et al., 2013). It is particularly interesting that Guo et al demonstrated a similar effect of AKT on GSK3β and also identified eIF2Bɛ as an important downstream effector of GSK3β in axon regeneration (Guo et al., 2016). mTORC1 inhibition (deletion of Rptor or Mtor, over-expression of dominant negative mutant S6K1-DN or 4E-BP1-4A) decreased AKT-induced ON regeneration (Miao et al., 2016), consistent with our conclusion that mTORC1 is necessary for axon regeneration. Therefore, work from several laboratories, including our own, concurs that mTORC1 activation and GSK3β inhibition act in parallel and synergistically downstream of AKT to promote CNS axon regeneration (Guo et al., 2016; Miao et al., 2016).
AKT inhibition significantly reduces PTEN deletion-induced ON regeneration
To definitively determine the role of AKT in PTEN deletion-induced axon regeneration, we crossed PTEN floxed mice with AKT3 knockout (KO) mice to generate conditional PTEN/AKT3 double KO (DKO) mice. Intravitreal injection of AAV2-Cre + AAV-U6-AKT1/2 shRNA in this mouse line to inhibit the three isoforms of AKT significantly reduces, but does not completely abolish, PTEN KO-induced ON regeneration (Huang et al., 2019). Thus it seems that AKT is both necessary and sufficient to promote ON regeneration. However, it is still not clear whether AKT is the sole effector downstream of PTEN for axon regeneration.
Marginal AKT activation after PTEN deletion due to mTORC1-S6K1 feedback inhibition in RGCs results in adequate mTORC1 activation and GSK3β phosphorylation/inhibition for ON regeneration
Our investigations provided additional details about the activity and influence of AKT in PTEN KO mice. We found that AKT phosphorylation (pAKT) is only slightly increased, but mTORC1 is activated robustly, in the RGCs with PTEN deletion (Huang et al., 2019), consistent with previous reports in non-neuronal systems that mTORC1 limits AKT activation through feedback inhibition (Fig. 1) (Laplante & Sabatini, 2012; L. Yang et al., 2014). We also demonstrated that abolishing mTORC1 activity by deletion of both Rptor and Pten significantly decreases pS6 but increases AKT phosphorylation, while significantly decreasing axon regeneration. This result indicates a feedback loop in PTEN KO RGCs that tightly controls the activity of AKT (Huang et al., 2019). Overexpressing S6K1 in PTEN KO mice further decreases AKT phosphorylation and reduces axon regeneration (L. Yang et al., 2014). Possibly through a similar mechanism, S6K inhibits axon regeneration in C. elegans (Hubert, Wu, Chisholm, & Jin, 2014) and inhibition of S6K1 promotes corticospinal tract regeneration in mice (Al-Ali et al., 2017). We also found that GSK3β-S9A overexpression significantly decreases PTEN KO-induced axon regeneration, and that combining Rptor deletion and GSK3β-S9A overexpression further decreases without totally abolishing PTEN KO-induced axon regeneration, confirming the synergistic effects of these two parallel downstream effectors of AKT (Miao et al., 2016). The balance between PTEN KO forward activation and mTORC1 feedback inhibition therefore limits AKT activation, which suggests that AKT serves more of a necessary function since it induces adequate mTORC1 activation and GSK3β inhibition, which are essential for axon regeneration. It would not be surprising if additional balancing mechanisms can fine-tune the growth control loop of PI3K-AKT-mTORC1-PI3K. This feedback inhibition minimizes AKT activation even after PTEN deletion (Laplante & Sabatini, 2012; L. Yang et al., 2014), suggesting that AKT-independent signals downstream of PTEN contribute to axon regeneration. PTEN deletion-induced PIP3-dependent signaling includes many AKT-independent pathways (Lien, Dibble, & Toker, 2017). In addition, PTEN can dephosphorylate focal adhesion kinase (FAK) and Shc, and deletion of PTEN activates FAK, RAS and ERK (Godena & Ning, 2017). Furthermore, PTEN in the nucleus also plays a non-catalytic role in chromosomal instability, DNA repair, and pre-mRNA alternative splicing (S. M. Shen et al., 2018; W. H. Shen et al., 2007; Song et al., 2011). Elucidation of these PTEN-dependent but AKT-independent pathways in axon regeneration will be an important future direction for the field.
Forced AKT activation or GSK3β inhibition enhances ON regeneration in Pten KO mice, suggesting synergism between AKT-dependent and AKT-independent pathways downstream of PTEN
We used AAV-mediated AKT overexpression to overcome the feedback inhibition of mTORC1 and therefore to force AKT activation in PTEN KO mice. As expected, intravitreal injection of AAV2-myr-AKT3 in PTEN KO mice significantly increases phosphorylation levels of AKT and GSK3β; and generates significantly more and longer regenerating axons than PTEN KO alone. AAV2-Cre mediated Pten/Gsk3β DKO also produces more potent axon regeneration than PTEN KO alone (Huang et al., 2019). Fully activated AKT or total deletion of GSK3β therefore acts synergistically with AKT-independent signaling downstream of PTEN deletion to promote CNS axon regeneration.
Conclusion and future perspective
By applying AAV to the mouse RGC/ON system to achieve an extensive molecular dissection of the PTEN/PI3K/AKT/mTORC1/mTORC2/GSK3β pathways specifically in RGCs, we have greatly enhanced understanding of the mechanisms by which PTEN regulates neuron-intrinsic regrowth ability and definitively identified the linear and parallel signals downstream of PTEN that contribute to ON regeneration. The results raise intriguing questions that must be answered before a safe and effective regenerative therapy can be developed. For example, our findings that: 1) mTORC1 is necessary for ON regeneration and 2) that mTORC2 is inhibitory for AKT-induced axon regeneration (Miao et al., 2016; L. Yang et al., 2014) imply that modulation of the delicate balance between mTORC1 and mTORC2 may optimize ON regeneration. mTORC2 and pAKT-S473 are necessary for PTEN deletion-induced tissue overgrowth in mouse prostate cancer (Guertin et al., 2009) and drosophila eyes (Hietakangas & Cohen, 2007). Therefore blocking mTORC2 and pAKT-S473 in PTEN KO mice may allow us to minimize their deleterious tumorigenic effect while boosting PTEN/AKT’s regeneration-promoting effect. The increased understanding of the complicated cross-regulation and feedback-control mechanisms involved in mTORC1/2 will certainly inspire more studies on these critical growth control mechanisms, which will eventually lead to safe and effective therapeutic strategies for CNS injury that avoid the molecular targets that mediate deleterious effects. We also found that: 3) Slight AKT activation after PTEN deletion produces adequate mTORC1 activation and GSK3β phosphorylation/inhibition for axons to regenerate. Clearly, however, these low levels of AKT activation and GSK3β inhibition themselves are insufficient for axon regeneration. Therefore, partially activated AKT plays a necessary role in PTEN deletion-induced ON regeneration. 4) mTORC1 activation by TSC deletion (Park et al., 2008), constitutively active mutant of S6K1 (L. Yang et al., 2014) or AKT overexpression (Miao et al., 2016) can induce axon regeneration, but to a much lesser degree than PTEN deletion. These data also argue that increase of global translation is necessary but not sufficient for potent axon regeneration. A more specific permissive signal may be critical to initiate axon regeneration. 5) Combining AKT activation with PTEN deletion has a significant synergistic effect in axon regeneration. All of these results suggest that AKT is a downstream effector of PTEN but cannot fully account for the effect of PTEN deletion on axon regeneration. An AKT-independent pathway downstream of PTEN is therefore the key to ignite and propel neuron-intrinsic growth machinery proactively, whereas AKT-dependent signaling is necessary and supportive for axon regeneration (Fig. 1). It is extremely intriguing and important to identify these permissive and proactive signals of axon regeneration and elucidate the mechanisms by which they are cross regulated with the necessary AKT-mTORC1 signals to release the full potential of PTEN deletion in ON regeneration. This is a critical step toward developing safe and effective strategies for CNS repair.
Conflict of interest statement
The authors have declared that no conflict of interest exists.
Footnotes
Acknowledgments
This work was supported by grants from NIH NEI (EY024932, EY023295 and EY028106) to YH, and partially supported by Research for Prevention of Blindness Unrestricted grant and NEI P30-EY026877.