
Abstract
Background:
Traumatic spinal cord injury (SCI) is a complex medical condition causing significant physical disability and psychological distress. While the adult spinal cord is characterized by poor regenerative potential, some recovery of neurological function is still possible through activation of neural plasticity mechanisms. We still have limited knowledge about the activation of these mechanisms in the different stages after human SCI.
Objective:
In this review, we discuss the potential role of biomarkers of SCI as indicators of the plasticity mechanisms at work during the different phases of SCI.
Methods:
An extensive review of literature related to SCI pathophysiology, neural plasticity and humoral biomarkers was conducted by consulting the PubMed database. Research and review articles from SCI animal models and SCI clinical trials published in English until January 2021 were reviewed. The selection of candidates for humoral biomarkers of plasticity after SCI was based on the following criteria: 1) strong evidence supporting involvement in neural plasticity (mandatory); 2) evidence supporting altered expression after SCI (optional).
Results:
Based on selected findings, we identified two main groups of potential humoral biomarkers of neural plasticity after SCI: 1) neurotrophic factors including: Brain derived neurotrophic factor (BDNF), Nerve growth factor (NGF), Neurotrofin-3 (NT-3), and Insulin-like growth factor 1 (IGF-1); 2) other factors including: Tumor necrosis factor-alpha (TNF-α), Matrix Metalloproteinases (MMPs), and MicroRNAs (miRNAs). Plasticity changes associated with these biomarkers often can be both adaptive (promoting functional improvement) and maladaptive. This dual role seems to be influenced by their concentrations and time-window during SCI.
Conclusions:
Further studies of dynamics of biomarkers across the stages of SCI are necessary to elucidate the way in which they reflect the remodeling of neural pathways. A better knowledge about the mechanisms underlying plasticity could guide the selection of more appropriate therapeutic strategies to enhance positive spinal network reorganization.
List of abbreviations
Spinal cord injury
Blood spinal cord barrier
α TNF alpha
Central nervous system
Transcranial magnetic stimulation
Functional magnetic resonance imaging
Cerebrospinal fluid
Brain derived neurotrophic factor
p75 neurotrophin receptor
Mature BDNF
Matrix metalloproteinase-9
Tropomyosin receptor kinase B
Long-term potentiation
N-methyl-D-aspartate
Gamma-aminobutyric acid
Potassium-chlorine cotransporter
Nerve growth factor
Peripheral nervous system
Tropomyosin receptor kinase A
Mature NGF
Matrix metalloproteinase-7
Endoplasmic reticulum
C/EBP homologous transcription factor
Glucose-regulated protein 78
Growth-associated protein 43
Neuron-specific enolase
Glial-derived neurotrophic factor
American Spinal Injury Association
Functional Independence Measure
Neurotrofin-3
Mature NT-3
Tropomyosin receptor kinase C
Corticospinal tract
Insulin-like growth factor 1
Growth hormone
IGF-1 receptor
Mitogen-activated protein
Phosphoinositide 3-kinase
Traumatic brain injury
Messenger RNA
ASIA impairment scale
TNF-α-converting enzyme
TNF receptor-associated death domain
Nuclear factor kappa B
c-Jun N-terminal kinase
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate
Matrix Metalloproteinases
Extracellular matrix
Tissue inhibitors of metalloproteinases
Fas ligand
MicroRNAs
Phosphatase and tensin homolog
Mechanistic target of rapamycin
Blood brain barrier
Glial fibrillary acid protein
Dynamic contrast-enhanced magnetic resonance imaging.
Introduction
Traumatic Spinal Cord Injury (SCI) is an invalidating medical condition resulting from a disruption of neural signaling through afferent and efferent pathways and loss of information processing within the damaged region of the spinal cord. From a pathophysiological point of view, SCI can be divided into four progressive stages: physical trauma, primary injury, secondary injury and formation of the glial scar (Tran et al., 2018). Physical trauma provokes the primary injury of spinal cord tissues leading to local cell death. Vascular disruption occurs in parallel, resulting in hemorrhage, vasospasm, additional reperfusion injury, and breakdown of the blood-spinal cord-barrier (BSCB). The response of the remaining viable neural tissue, surrounding the damaged spinal cord, leads to secondary injury, a condition marked by intense neuroinflammation. An underlying cascade of molecular events results in the spread of neuronal and glial necrosis and apoptosis beyond the initial site of trauma and in the formation of the glial scar. The glial scar is a physiological attempt to stabilize the expansion of secondary injury, but, at the same time, it has a deleterious effect on neural regeneration acting as a physical and molecular barrier for axonal growth (Ahuja et al., 2017). Recent studies indicate that the function of the glial scar extends beyond the inhibition of axonal growth. Both infiltrating immune cells and resident glia undergo structural and functional reorganization after SCI enabling them to release pro-inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α) and interferon gamma, interleukins and chemokines (Popovich et al., 1997). These humoral factors have various pathophysiological functions in the central nervous system (CNS) and many of them have been shown to have a deleterious effect on functional repair after SCI.
Parallel to these mainly detrimental events, the CNS activates intrinsic mechanisms of repair in order to regain or compensate the loss of neurological functions. To some extent, the spontaneous recovery of sensorimotor functions is possible within the first few months after SCI due to the resolution of neuropraxia and remyelination of spared axons (Curt et al., 2008; Dietz & Fouad, 2014). However, probably the most important and sophisticated factor that guides long-term functional recovery during the process of neurorehabilitation is neuroplasticity, i.e. the ability of the CNS to undergo structural and functional changes in response to SCI. Neuroplasticity after SCI occurs at several anatomical levels of the CNS, including the spinal cord above and below the level of injury (Grau et al., 2014), the brainstem and the sensory-motor cortex (Bruehlmeier et al., 1998; Freund et al., 2013; Jurkiewicz et al., 2007; Onifer et al., 2011). It includes alterations of intracellular properties (Boulenguez et al., 2010; Murray et al., 2010), strengthening or weakening of pre-existing synapses (Rioult-Pedotti et al., 2007), activation of silent connections, and axonal sprouting with formation of new neural circuits (Bareyre et al., 2004). The activated mechanisms underlying neuroplasticity depend on the time-frame of SCI and are strongly influenced by the levels of inflammation and rehabilitation training-induced neural activity. The outcome of this complex interplay is a series of both adaptive and maladaptive changes within the spinal cord networks that can lead to functional improvement of the neurological deficit or, on the contrary, to the establishment of SCI-related complications (Ferguson et al., 2012; Grau et al., 2014).
Understanding how and to what extent certain aspects of neuroplasticity after SCI contribute to functional recovery is fundamental for the identification and adoption of strategies to strengthen the adaptive (as opposed to maladaptive) plasticity. Neuroplasticity-induced alterations of the CNS after SCI may be assessed by various approaches revealing changes at the molecular, cellular, structural, functional, and behavioral level. The final neurological outcome after SCI is measured in terms of clinical function and can be determined by evaluating the neurological status and electrophysiological recordings (Curt et al., 1998; Hupp et al., 2018; Lewko et al., 1995). CNS functional and structural reorganization and connectivity after human SCI can be assessed by transcranial magnetic stimulation (TMS) and functional magnetic resonance imaging (fMRI) (Levy et al., 1990; Topka et al., 1991). Direct evaluation of changes at the cellular and molecular level in human experimental studies remains still challenging for technical and ethical reasons. However, since the permeability of the BSCB is altered, SCI-induced factors involved in modulation of spinal cord plasticity, such as cytokines and growth factors, may be measured indirectly in the cerebrospinal fluid (CSF) and blood using a wide spectrum of highly sensitive immunoassays able to detect subtle longitudinal changes (Badhiwala et al., 2018; Kwon et al., 2011; Kwon et al., 2017).
Humoral biomarkers of SCI have previously been investigated during the acute phase as possible indicators of severity and early predictors of prognosis (Ahadi et al., 2015), but their application during the later phases of SCI has been less studied. Serial evaluation of humoral biomarkers may provide important information about individual responses to SCI. This approach may help to optimize and personalize therapeutic strategies through the identification of the activated neural plasticity mechanisms before and during the course of treatment. In the present review, we will discuss the possible role of various biomarkers of SCI as indicators of the plasticity mechanisms at work during the different phases of SCI (see Tables 1 2). An extensive review of literature related to SCI pathophysiology, neural plasticity and humoral biomarkers was conducted by consulting the PubMed database. Research and review articles from both, SCI animal models and SCI clinical trials published in English until January 2021 were reviewed. The selection of candidates for humoral biomarkers of plasticity after SCI was based on the following criteria: 1) evidence supporting altered expression after SCI; 2) evidence supporting involvement in neural plasticity.
Growth factors as candidates for SCI neuroplasticity biomarkers
Growth factors as candidates for SCI neuroplasticity biomarkers
As suggested by Kwon et al. 2019, temporal profile is expressed using the following definitions: superacute: < 6 h; acute: 6–48 h; subacute: 3–10 days; chronic > 10 days. Table summarizes a list of publications providing data from animal models and clinical trials that are specific to studies that look at biomarkers for neuroplasticity in different or in multiple phases of SCI.
Other factors as candidates for SCI neuroplasticity biomarkers
As suggested by Kwon et al. 2019, temporal profile is expressed using the following definitions: superacute: < 6 h; acute: 6–48 h; subacute: 3–10 days; chronic > 10 days. Table summarizes a list of publications providing data from animal models and clinical trials that are specific to studies that look at biomarkers for neuroplasticity in different or in multiple phases of SCI.
Brain derived neurotrophic factor
Brain derived neurotrophic factor (BDNF) is one of the most well-investigated modulators of neural plasticity mechanisms. BDNF is a member of the neurotrophic family of growth factors and exerts diverse modulatory actions through activation of distinct signaling pathways. Physiologically, there are two active forms of BDNF that play an important role in several functions (Lu, 2003; Lu et al., 2005) and exert even opposing effects depending on the activated receptor. The precursor protein, pro-BDNF, an immature form, mainly activates p75 neurotrophin receptor (p75NTR), a low-affinity nonselective neurotrophic receptor, and produces a variety of cellular events ranging from neuronal differentiation to apoptotic cell death (Kenchappa et al., 2006; Koshimizu et al., 2009; Teng et al., 2005; Yang et al., 2014). Mature BDNF (mBDNF) is generated by cleavage of pro-BDNF by extracellular proteases, such as matrix metalloproteinase-9 (MMP-9) and plasmin (Ethell & Ethell, 2007; Hwang et al., 2005). mBDNF modulates cellular functions mainly through its high-affinity ligand-specific receptor, tropomyosin receptor kinase B (TrkB), and plays an important role in neuronal development, synaptic transmission, cellular and synaptic plasticity as well as in the formation and maintenance of certain forms of memory (Kowiański et al., 2018).
BDNF also plays an important role in synaptic plasticity through the modulation of long-term potentiation (LTP). LTP, defined as an activity-dependent prolonged increase of synaptic efficacy, is the leading experimental model for experience-induced synaptic changes (Malenka & Nicoll, 1999). In general, LTP induction and maintenance require intact BDNF signaling between different cellular substrates such as the N-methyl-D-aspartate (NMDA) receptor, signaling kinases, post-translational modification and transcription. Since LTP is believed to be a neural substrate of learning and memory, it has mostly been studied and best described in the hippocampus. However, evidence suggests that it is a widespread phenomenon expressed at all excitatory synapses (Malenka & Bear, 2004), including those in the spinal cord. Indeed, it has been demonstrated that spinal cord neural circuits have the capacity to adapt in response to training, peripheral stimulation and injury through BDNF-mediated mechanisms. In the intact spinal cord, exercise and instrumental training may promote neural plasticity and learning by up-regulation of endogenous BDNF expression (Baumbauer et al., 2009; Gómez-Pinilla et al., 2007; Gómez-Pinilla et al., 2001; Huie, 2012). In pathological conditions, such as hypoxia, BDNF may induce adaptive functional modifications within the spinal cord (Baker-Herman et al., 2004; Dale-Nagle et al., 2010), while after SCI it promotes recovery of locomotor function (Boyce et al., 2012; Boyce et al., 2007) and counters maladaptive plasticity restoring the capacity for learning (Grau et al., 2014).
Beside these beneficial effects, multiple evidence shows that BDNF signaling in the spinal cord promotes nociception. Since TrkB is expressed on the dorsal horn neurons, a group of neurons receiving nociceptive afferents, it is reasonable to postulate that BDNF is involved in the modulation of pain perception. It has been shown that, in the absence of SCI, both inflammatory and chronic neuropathic pain are modulated by BDNF through a variety of mechanisms, including increase in neural excitation (Thompson et al., 1999), phenotypic switch of BDNF expressing neurons (Ohtori et al., 2002), upregulation of BDNF expression in sensory neurons (Fukuoka et al., 2001; Ha et al., 2001; Miletic & Miletic, 2002) and LTP-like synaptic modification of excitatory responses in lamina II neurons leading to central sensitization (Garraway et al., 2005). These effects may be related to altered regulation of intracellular Cl– concentrations and the switch of gamma-aminobutyric acid (GABA) function from inhibitory to excitatory. High concentrations of BDNF have been shown to reduce the expression of the membrane-bound potassium-chlorine cotransporter (KCC2). In turn, the intracellular concentration of Cl– remains high and GABAA receptor chloride ion channels allow Cl– to leave the cell, causing depolarization and promoting neural excitation (Zhang et al., 2013).
Since neuropathic pain is a frequent complication of SCI, the apparent shift of BDNF function from mediation of adaptive plasticity to maladaptive alteration of neural circuits has overshadowed its potential application as a biomarker of SCI. Indeed, despite the important restorative potential of BDNF, no clinical trial has yet investigated the temporal dynamics of BDNF signaling across different stages of SCI. Garraway and colleagues (2016) shed some light on the complex role of BDNF, arguing that BDNF probably exerts a dual function in SCI. In the acute phase, reduced levels of BDNF produce maladaptive spinal plasticity and impair functional recovery. However, during the chronic phase, when BDNF levels have been restored and TrkB expression is increased, even a minimal overexpression of BDNF may induce maladaptive plasticity leading to central sensitization and development of chronic neuropathic pain condition. Therefore, it would be important to investigate the alterations of BDNF signaling during the different phases of SCI to better understand its role in modulating plasticity-driven restorative potential and the emergence of complications, such as neuropathic pain. Given the opposing functions of pro-BDNF and mBDNF, it would also be of great interest to study the concentration of these two forms of BDNF as well as the mechanisms involved in the cleavage of pro-BDNF to mBDNF after SCI. This information would enable clinicians to select the optimal therapeutic strategy at each different SCI time-point to promote adaptive recovery mechanisms and prevent the onset of neuropathic pain, based on the individual patient BDNF profile.
Nerve growth factor
During development, nerve growth factor (NGF) is involved in the regulation of neuronal growth, differentiation, and survival in the peripheral nervous system (PNS) (Heumann et al., 1987). NGF also plays an important neuroprotective role in the adult CNS by modulating the neural responses to injury of cell types that express NGF receptors (Connor & Dragunow, 1998). NGF mediates protective effects mainly through activation of ligand-specific tropomyosin receptor kinase A (TrkA) by its mature form (mNGF) (Levi-Montalcini, 1987). In contrast, pro-NGF, a precursor of NGF, binds preferentially to p75NTR that, as mentioned above, exerts opposite biological functions leading to apoptosis. Pro-NGF is processed to NGF intracellularly by action of furin convertase (Seidah et al., 1996) or extracellularly by action of proteases plasmin and matrix metalloprotease-7 (MMP-7) (Bruno & Cuello, 2006; Lee et al., 2001; Smith et al., 1995). Since pro-NGF is the most abundant NGF form in the brain, it is feasible to surmise that the conversion of pro-NGF into mNGF may be critical in determining the extent of cell death and reparation after injury. Alteration of MMP-7 enzymatic activity leads to additional incrementation of pro-NGF and p75NTR signaling, favoring degenerative processes. Indeed, enhanced pro-NGF signaling has been detected in the brain of Alzheimer disease patients (Peng et al., 2004), in the hippocampus after seizure (Volosin et al., 2008) and after SCI (M. S. Beattie et al., 2002).
Under normal conditions, only a small amount of NGF is found in the spinal cord. Nevertheless, NGF protein levels rise after injury at the damaged spinal cord level (Bakhit et al., 1991) and may be involved in structural plasticity, guiding regenerating axons across the lesion site and promoting the regenerative response (Tuszynski et al., 1994). The neuroprotective role of NGF may be related to the inhibition of endoplasmic reticulum (ER) stress-induced apoptotic cell death via the activation of downstream signals (Kishi et al., 2010). Some of the events triggered by the secondary injury, such as accumulation of unfolded proteins in the ER lumen, inhibition of protein synthesis, depletion of Ca2 + from ER stores, and activation of the expression of C/EBP homologous protein (CHOP) transcription factor, glucose-regulated protein 78 (GRP78) and caspase-12, are involved in neuronal cell apoptosis signaling after SCI (Ohri et al., 2011; Soboloff & Berger, 2002; Valenzuela et al., 2012). Zhang et al. (2014) demonstrated that the exogenous administration of NGF in an animal model of SCI had indeed a protective effect by acting on ER function after injury. This effect was mediated by decreased levels of CHOP, GRP78 and caspase-12 and increased expression of growth-associated protein 43 (GAP43), which prevented apoptosis, promoting adaptive plasticity and recovery of function.
Structural plasticity alterations mediated by NGF signaling within spinal cord circuits after SCI may be also maladaptive. Beside neurons, glial and meningeal cells have been identified as additional sources of intraspinal NGF after SCI contributing to the enhancement of its levels (Brown et al., 2004). Increased levels of NGF after injury induce robust sprouting of unmyelinated afferent C-fibers and lightly myelinated afferent Aδ-fibers, thereby contributing to the development of pain-related behaviors and autonomic dysfunction. There is evidence that strategies that counteract NGF signaling may prevent or ameliorate the effects of aberrant plasticity. Attenuation of mechanical hyperalgesia, a type of central pain syndrome associated with SCI, has been observed after a cycle of daily administration of antibodies to NGF in an animal model (Brown et al., 2004). In addition, treatments based on antibodies against NGF may modulate the onset of autonomic dysfunction, like dysreflexia. Dysreflexia is a direct consequence of the lesion of spinal pathways characterized by the absence of descending control of spinal reflexes. A typical consequence of this medical condition is the sudden increase in heart rate and arterial pressure after bladder or colon distension. Injury-induced plasticity leads to NGF-dependent enlargement of the central arbor of a sub-population of sensory neurons that provide increased afferent input to the spinal reflex, intensifying autonomic dysreflexia (Shelton & Reichardt, 1984). Indeed, it has been shown in animal studies that blocking intraspinal NGF with an intrathecally-delivered neutralizing antibody to NGF prevents small-diameter afferent sprouting, thereby decreasing significantly the symptoms of dysreflexia (Krenz et al., 1999).
The possible involvement of NGF signaling in the control of bladder function under physiological and SCI conditions in both the CNS and PNS is an additional important area of study in the field of SCI. Complete SCI, rostral to the lumbosacral spinal cord, irreversibly affects neural circuitry involved in urine storage and elimination leading to detrusor sphincter dyssynergia and detrusor overactivity (Tai et al., 2006). It has been demonstrated that neutralizing NGF action in the lumbosacral spinal cord with NGF-specific antibodies prevents the onset of both medical conditions in animal models (Seki et al., 2002; Seki et al., 2004). However, since it has been shown that pro-NGF, but not mNGF, is released in the cord after SCI (M. S. Beattie et al., 2002), one may postulate that the pathway silenced by the antibody is actually the pro-NGF/p75 one, given that the two domains are shared. In support of this, it has been recently shown that pharmacological blockage of pro-NGF/p75 interaction restores bladder function (Ryu et al., 2018). Additionally, NGF has been investigated as a possible urine biomarker of bladder dysfunction, since it is primarily secreted by the urinary bladder mesothelium and transported in retrograde fashion to the sacral micturition centers (Vizzard, 2006). Of note, urinary levels of NGF were found to be higher in patients with SCI who developed elevated intravesical pressure due to neurological bladder dysfunction than in adult controls. Also, in a clinical study on patients with overactive bladder, the urinary NGF/Creatinine ratio showed a difference between urinary retention states after catheterization and relief of increased pressure, which indicates that urinary NGF levels may also be a valid biomarker of treatment responses (Kuo et al., 2010).
Despite multiple evidence showing the involvement of NGF in the regulation of functional recovery after SCI, so far only one clinical study has investigated the alteration of NGF expression: de Mello Rieder et al. (2019) conducted a prospective cohort study over a period of 3 years on 52 patients with SCI due to traffic accidents, falls and firearm wounds and 36 healthy subjects with no history of SCI as a control group. Blood samples were collected within 48 h and at 7 days after SCI and neuron-specific enolase (NSE), interleukin-6, glial-derived neurotrophic factor (GDNF) and NGF were measured to evaluate an acute-phase reactive cellular response to traumatic SCI. Compared to the control group, the concentration of NGF was significantly lower at both time-points, 48 h and 7 days after SCI, and no significant correlation was found with neurological outcomes as measured by American Spinal Injury Association (ASIA) and Functional Independence Measure (FIM) scores. These data demonstrate suppression of NGF expression and decreased neurotrophic support during the acute phase of SCI generally characterized by sustained neural cell death. It would be interesting to explore if there is an alteration of the conversion of pro-NGF into mNGF during this early phase that may contribute to the decrease in mNGF concentration and to the increase in pro-NGF signaling, ultimately leading to apoptotic cell death of neurons indirectly affected by the injury and contributing to the expansion of secondary injury. However, considering the preliminary nature of this study and the small sample size, additional clinical trials are needed to confirm these data. Moreover, it would be important to follow the progression of this initial NGF suppression during the successive phases of SCI and to correlate eventual different NGF expression profiles with adaptive and maladaptive functional outcomes.
Neurotrofin-3
Neurotrofin-3 (NT-3) is a third member of the neurotrophic factor family. It was detected in glia and neuronal populations in the CNS, with higher concentrations during development than in the adult period, indicating its important role in the regulation of neuronal survival and differentiation during the early phases of life (Ernfors et al., 1990; Maisonpierre, 1990). Its role as a trophic factor is particularly important for the growth of several groups of neurons, such as sympathetic, sensory and motor neurons (Fariñas et al., 1994; Ringstedt et al., 1993). In a similar fashion to BDNF and NGF, the mature form of NT-3 (mNT-3) is obtained from its precursor peptide pro-NT-3 by proteolytic action of furin convertase (Farhadi et al., 2000). The principal trophic effects of mNT-3, including mitogenesis, promotion of survival, and differentiation, depend on the developmental stage of the target cells and are mainly mediated through high-affinity activation of ligand-specific tropomyosin receptor kinase C (TrkC) (Lamballe et al., 1991). On the other side, as reported for the other immature form of neurotrophic factors, pro-NT-3 binds preferentially to p75NTR receptor promoting apoptotic cell death. Notably, mNT-3 can also bind, with lower affinity, to TrkA and TrkB receptors, although the physiological role of this ligand-receptor interaction is unclear (Benedetti et al., 1993).
Of all neurotrophins, NT-3 is the one most expressed in the developing spinal cord, particularly in motor neurons, reflecting its putative role as an inducer and regulator of corticospinal tract (CST) collateral sprouting, an important step to target-finding and innervation (Ernfors & Persson, 1991; Friedman et al., 1991; Maisonpierre, Belluscio, Friedman, et al., 1990). Moreover, TrkC receptor is highly expressed in the deep layers of the developing cortex where the neurons programmed to form the descending CST originate (Ringstedt et al., 1993). The corresponding layers of the cortex in the adult continue to express TrkC receptors, although at lower levels, indicating that the CST may preserve its responsiveness to NT-3 in adulthood. Though NT-3 expression is minimal in the adult spinal cord under physiological conditions, there is strong evidence from animal studies that artificial expression of NT-3 after SCI at the site of lesion, via different techniques, promotes axonal plasticity and induces growth and sprouting of the CST fibers. Implantation of grafts containing NT-3 expressing cells at lesion sites stimulates CST growth over short distances, during both the acute (Grill et al., 1997; Ma et al., 2010; Shang et al., 2011) and chronic phases of SCI (Kusano et al., 2010; Tuszynski et al., 2003), enabling a modest amount of functional recovery of locomotor skills. In a similar way, viral vector-induced expression of NT-3 promotes regrowth of CST fibers when injected into the rostral spinal cord (Weishaupt et al., 2014) as well as collateral sprouting of CST axons across the midline towards denervated motor neurons expressing NT-3 (Chen et al., 2006; Zhou et al., 2003). Recently, a published study by Rao et al. (2018) reported that insertion of the biodegradable material chitosan loaded with NT-3 into a lesioned rhesus monkey thoracic spinal cord elicited robust long-distance axonal regeneration, thus confirming that NT-3 is a promising tool to promote rewiring of the CST also in primate CNS under pathological conditions. This study also evaluated a battery of outcome measures, including fMRI, magnetic resonance diffusion tensor imaging, and kinematics walking analyses, and found that neural regeneration induced by NT-3 chitosan was accompanied by sensory and motor functional recovery. The same group of authors had previously shown in a rodent model that the synergic action of NT-3, a growth-stimulating and chemo-attractive factor, and chitosan, a bioactive material with anti-inflammatory action, is critical for the successful regeneration of neural circuits, indicating the importance of the interplay between neuroplasticity and neuroinflammation for recovery after SCI (Yang et al., 2015).
While there is robust evidence from animal studies that NT-3 signaling plays a critical role in the reconnection between injured CST axons and spared neurons, little is known about the profile of its expression during the different phases of SCI. Since it has been shown in rats that over-expression of NT-3 promotes sprouting of CST axons in the injured, but not in the intact spinal cord, it has been hypothesized that processes associated with injury, such as Wallerian degeneration and inflammation-induced alterations of the micro-environment, participate in the modulation of NT-3 signaling, leading to the induction of neuroplasticity. Therefore, clinical trials designed to serially dose NT-3 at different stages of SCI may help to understand whether this neurotrophic factor could be a potential biomarker for evaluating the structural plasticity potential. In addition, given the promising results from animal studies regarding the over-expression of NT-3, measuring NT-3 after SCI may help detect those patients who could benefit most from the treatment. To date, there is no evidence of any association of NT-3 with maladaptive plasticity, such as chronic pain conditions or spasticity. This offers great advantages for treatment strategies based on enhanced NT-3 signaling and indicates their potential to facilitate meaningful recovery after SCI.
Insulin-like growth factor 1
Insulin-like growth factor 1 (IGF-1) is a polypeptide hormone involved in somatotropic axis signaling. The major source of circulating IGF-1 is the liver, where its expression and secretion are regulated by growth hormone (GH). After release, IGF-1 binds, both in the circulation and in tissues, to high affinity IGF1-binding proteins involved in modulating the interaction between IGF-1 and its receptor. The biological effects of IGF-1 are mediated through the activation of IGF-1 receptor (IGF1-R), a membrane-bound tyrosine kinase receptor. IGF1-R is ubiquitously expressed, and it acts via the mitogen-activated protein (MAP) kinase and phosphoinositide 3-kinase (PI3K) signaling pathways to promote tissue growth and maturation through upregulation of anabolic processes. Since IGF-1 shares part of its structure with insulin, it can act also through the insulin receptor. IGF-1 reaches the CNS from plasma through the choroid plexus via mechanisms of active transport (Carro et al., 2003; Santi et al., 2018) and it is also locally secreted by neurons and glial cells in a GH-independent manner, with microglial cells as the major source (Labandeira-Garcia et al., 2017; Quesada et al., 2007; Rodriguez-Perez et al., 2016; Suh et al., 2013). IGF-1 is involved in the regulation of multiple physiological processes in the CNS including brain development, myelination, synapse formation, synaptic transmission, adult neurogenesis and cognition (Nieto-Estévez et al., 2016; Wrigley et al., 2017).
There is growing evidence that IGF-1 signaling is involved in the modulation of neural function also under pathological conditions. Preclinical studies based on multiple animal models of a wide spectrum of pathologies affecting the CNS and sensory deprivation have demonstrated the involvement of IGF-1 in neural protection, reparation and plasticity (Wrigley et al., 2017). IGF-1 signaling is high in the CNS at early developmental stages and declines significantly across the lifespan (Werner et al., 1989). It has been shown that some forms of brain injury, such as cerebral ischemia and traumatic brain injury (TBI), induce upregulation of IGF-1 signaling (Bergstedt & Wieloch, 1993; Gluckman & Ambler, 1992; Madathil et al., 2010), probably as part of a neuroprotective response by damaged CNS tissue (Lee et al., 1996). Neuroprotective effects have also been shown when IGF-1 is supplied through different routes, such as direct intracerebroventricular administration (Brywe et al., 2005; Guan et al., 1993) or peripheral administration (Fernandez et al., 1999; Tagami et al., 1997). Protective effects are mainly due to antiapoptotic mechanisms (Brywe et al., 2005; Guan et al., 2003), since IGF-1 has been shown to promote neural survival and inhibit apoptosis both in vitro (Delaney et al., 1999; Russell et al., 1998; Takadera et al., 1999; Van Golen & Feldman, 2000) and in vivo (Guan et al., 1993). There is also evidence that IGF-1 gene delivery promotes CST neuronal survival after SCI (Hollis et al., 2009; Nakao et al., 2001), although this effect was not associated with significant functional recovery. Interestingly, Liu et al. (2017) have shown in a mouse model of SCI that post-lesional adenoviral vector-assisted coexpression of IGF-1 and osteopontin (a matricellular protein with diverse biological functions) in cortical neurons leads to robust CST regrowth and the recovery of CST-dependent motor performance after T10 lateral spinal hemisection. This suggests that osteopontin may increase neuronal sensitivity to IGF-1 through interaction with cell-surface proteins leading to enhanced mobilization of IGF1-R to the cell membrane and improved responsiveness to the ligand. The critical involvement of IGF-1 signaling in axonal regeneration and neurological recovery after SCI has also been confirmed by Joshi et al. (2015) in an interesting study investigating the mechanisms underlying the lack of a neuronal intrinsic regenerative potential after CNS axonal injury. The main reported finding is the identification of an inhibitory molecular triad composed of the ubiquitin ligases MDM4, MDM2 and the transcription factor p53, which functions as a protein complex restricting the axonal regeneration program. Disruption of this inhibitory network through conditional deletion of ligases or pharmacological modulation of molecular interactions in a mouse model of SCI resulted in activation of p53 and enhancement of IGF-1 signaling as a final downstream effector, leading to axonal sprouting and functional recovery.
Results from animal models of SCI have demonstrated that IGF-1 expression is altered during different phases post-injury. In a model of SCI induced by cryogenic lesion in the spinal cord of adult rats, it was shown that expression of IGF-1 messenger RNA (mRNA) and IGF-1 peptide in astrocytes starts increasing from 3–7 days after injury, reaching a maximum at 21–28 days and declining to near control levels at 56 days (Yao et al., 1995). A similar pattern of increasing IGF-1 levels, both at the lesion site and within the dorsal root ganglia whose axons transverse the lesion, was shown after experimentally induced spinal cord demyelination in adult rats (Hinks & Franklin, 1999). In this study, IGF-1 levels peaked at 10 days, a time when new myelin sheaths appear, and declined within 28 days. Data from these studies indicate that early production of IGF-1 after SCI might be involved in myelin regeneration, thus exerting an additional neuroprotective effect.
The role of IGF-1 in regulation of SCI-related molecular mechanisms has also been studied in patients. To date, two prospective observational studies have investigated serial serum IGF-1 levels over a 12-week period after admission to hospital (Ferbert et al., 2017; Moghaddam et al., 2016). Both trials included follow-up of patients who were divided into two groups based on the level of neurological damage assessed by the ASIA impairment scale (AIS): those with neurological remission (improvement in the AIS score) vs. those without neurological remission (no improvement in AIS score). The results from these studies are overlapping and demonstrate in both groups of patients constant levels of serum IGF-1 after traumatic SCI during the acute phase until the third day after trauma, followed by a gradual increase during the post-acute and intermediate phase. Importantly, the mean IGF-1 serum level was higher in patients with neurological remission indicating a possible influence of IGF-1 on neuroprotective processes, as demonstrated in animal studies. Since the patients with neurological improvement showed consistently increased IGF-1 levels, Moghaddam et al. (2016) also analyzed whether the serum levels in the first 24 hours after SCI could already predict the AIS score improvement. After adjusting for relevant co-variables, multivariate analysis demonstrated a low capacity of IGF-1 serum levels in the first 24 hours to predict neurological outcome. However, to better understand the role of IGF-1 during different phases of SCI there is a need for larger human studies specifically designed to evaluate the fluctuation of its serum levels and its correlations with functional recovery.
Other factors
Tumor necrosis factor-alpha
TNF-α is a potent systemic pro-inflammatory molecule that plays a key role in the activation of the immune system in response to numerous intrinsic and extrinsic insults. TNF-α is a protein anchored to the cell surface as a stable homotrimer (Pennica et al., 1984) and subsequently processed and released by action of the metalloprotease TNF-α-converting enzyme (TACE) (Kriegler et al., 1988). Both forms of TNF-α, cell membrane-associated and soluble, can interact with two corresponding receptors, p55 (TNF-RI) and p75 (TNF-RII), leading to activation of downstream signaling cascades (Chen & Goeddel, 2002). In the CNS, astrocytes, microglia, and neurons represent both the major source of TNF-α and the principal target cells, since TNF-RI e TNF-RII are expressed on the surface of all three cell types (Kinouchi et al., 1991). Binding of TNF-α to its receptors can initiate apoptosis through activation of the TNF receptor-associated death domain (TRADD) or activate the nuclear factor kappa B (NFκB) and c-Jun N-terminal kinase (JNK) signaling pathways, both involved in activation/repression of key transcriptional targets (Park & Bowers, 2010). While TNF-α is one of the key factors driving the neuroinflammatory response provoked by neural tissue damage through activation of its major signaling pathways (Frankola et al., 2011), this cytokine is also involved in the modulation of various physiological processes in the CNS such as neuronal development, cell viability, neuronal ionic homeostasis, synaptic transmission, and synaptic plasticity (Butler et al., 2004; Chen & Goeddel, 2002; Cunningham et al., 1996; Schneider-Brachert et al., 2004).
Electrophysiological experiments performed on animal models have elucidated the effects of TNF-α signaling on synaptic plasticity (Ferguson et al., 2012; Grau et al., 2014). It has been shown that TNF-α leads to enhancement of spinal LTP in rats with neuropathic pain (Liu et al., 2007), indicating a possible role for TNF-α in modulating nociception. Indeed, it has been demonstrated that TNF-α is a key factor in the development of central sensitization and maladaptive plasticity believed to be the main underlying mechanisms of neuropathic pain (Czeschik et al., 2008; Park et al., 2011). These effects have been linked to the capacity of TNF-α to enhance the trafficking of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors (AMPARs) to the postsynaptic membrane (E. C. Beattie et al., 2002), leading to enhanced Ca2 + permeable GluR2-lacking AMPARs and increased postsynaptic excitability. The same TNF-α driven mechanisms could potentially lead to excitotoxicity after the SCI-induced neuroinflammatory response, leading to increased secondary cell death (Ferguson et al., 2008). Further research has provided evidence that TNF-α also mediates maladaptive plasticity in the spinal motor learning paradigm (Huie, 2012), since intrathecal delivery of TNF-α was found to be sufficient to compromise spinal learning. Interestingly, uncontrollable peripheral stimulation led to increased TNF-α release in the spinal cord, providing additional evidence that TNF-α signaling mediates stimulation-induced maladaptive plasticity. Inhibitors of TNF-α signaling pathway counteracted these effects, indicating that TNF-α plays a central role in the modulation of synaptic mechanisms of spinal cord learning.
TNF-α expression is upregulated after SCI (Haya-shi et al., 2000; Streit et al., 1998; Taupin et al., 1993; Wang et al., 1996; Wang et al., 2002; Yan et al., 2001), likely due to simultaneous production, tri-ggered by tissue damage, from different cell types including microglia, reactive astrocytes, neurons, endothelial cells, and infiltrating macrophages (Benveniste, 1992; Eng et al., 1992; Knoblach et al., 1999; Yan et al., 2001). A rapid and massive increase in TNF-α expression is evident within the first hours following SCI and it remains elevated for days (Wang et al., 1996). The peak of TNF-α in the acute phase of SCI promotes apoptotic cell death (Chi et al., 2008; Yune et al., 2003) and a local inflammatory reaction with damage of endothelium, spinal cord ischemia and edema (Carlos & Harlan, 1994; Rosenberg et al., 1995), aggravating the primary injury in this early stage of trauma. But contrary to the described deleterious effects in the acute phase of SCI, there is evidence suggesting that TNF-α signaling is beneficial during the chronic phase of SCI. The protective effect of TNF-α is mediated by the activation and proliferation of astrocytes involved in the restoration of the extracellular ion homeostasis, the modulation of neurotransmitter release and the production of neural growth factors, extracellular matrix molecules and adhesion molecules, all playing an important role in the modulation of neuroplasticity (Chi et al., 2008). TNF-α also upregulates calbindin and manganese superoxide dismutase (Bruce et al., 1996; Mattson et al., 1995), and promotes regeneration of injured CNS axons in vivo (Schwartz et al., 1991), additionally contributing to spinal cord recovery. This dual role of TNF-α has also been detected in the pathophysiology of TBI (Frankola et al., 2011).
The involvement of TNF-α in the pathophysiology of SCI has been studied also in humans. Two clinical studies evaluated the single shot level of TNF-α in heterogenous groups of post-acute patients, at different stages and severity of SCI, with respect to control groups. Both investigations reported an elevated serum level of TNF-α in the SCI group (Davies et al., 2007; Hayes et al., 2002). A more marked increase of TNF-α concentrations was registered in SCI patients with medical complications, such as chronic pain, recurrent urinary tract infections and/or pressure ulcers, indicating that peripheral inflammation probably modulates the global serum levels of TNF-α (Davies et al., 2007). The relationship between TNF-α and chronic pain condition in patients with SCI was further investigated in a study on 140 patients: 70 who developed neuropathic pain after injury vs. 70 controls who did not show neuropathic symptoms (Xu et al., 2015). Increased concentrations of TNF-α in serum were found to be associated with an elevated risk of developing neuropathic pain, indicating that TNF-α has a high potential to predict the onset of neuropathy and could become a diagnostic marker for this medical complication accompanying SCI. Since evidence from animal studies indicates that TNF-α is expressed within a few hours after injury, it has been also evaluated whether TNF-α may be an early clinical indicator of SCI severity. Temporal changes in TNF-α serum levels were measured in a pilot study including serial measurements during 12 weeks of follow-up in 23 patients after acute traumatic SCI with and without neurological improvement (Biglari et al., 2015). It was found that patients with neurological improvement had significantly lower TNF-α levels within the first hours post-injury, that then increased after 72h to the same levels as the group without improvement, and remained elevated also at later time points of the study. The higher TNF-α levels found in SCI patients with neurological improvement at later time points suggest that TNF-α is not necessarily associated only with exacerbation of neurological outcome, and that its role in the modulation of SCI depends not only on tissue and serum concentrations but also on the timing of SCI, as indicated by the animal studies discussed above. Due to the small sample size, these results have limited prognostic value, but are promising, and imply that further studies measuring the TNF-α expression profile after SCI may be meaningful for a better understanding of the role that this cytokine plays in limited behavioral recovery after injury, and hence for the development of specific treatment approaches.
Matrix Metalloproteinases
Matrix metalloproteinases (MMPs) are a vast group of proteolytic enzymes widely expressed in mammals, best known for their ability to regulate the extracellular matrix (ECM) dynamics (Shapiro, 1998). MMPs are expressed in an inactive form that requires further enzymatic processing to reveal the catalytic site and become active (Ethell & Ethell, 2007). Once activated, they are mainly inhibited by tissue inhibitors of metalloproteinases (TIMPs), but other processes such as glycosylation and internalization may also be involved. Balance between activation and deactivation is a critical step in the correct functioning of MMPs, and disruption of this homeostasis is observed in many pathological conditions. MMPs substrates include protein components of the ECM as well as growth factors, cytokines, chemokines, tyrosine kinase receptors, and cell adhesion molecules. Through cleavage of its targets, MMPs are involved in the regulation of multiple biological processes including cellular differentiation, migration, regulation of growth factor activity, survival or apoptosis, angiogenesis, inflammation, and signaling (Page-McCaw et al., 2007; Yong, 2005). In the CNS, MMPs also play an important role in the regulation of multiple aspects of neuronal physiology. The relatively high levels of MMPs expression during mammalian CNS development are associated with neurogenesis, neurite outgrowth, migration of neurons, axon guidance and myelination, topographic mapping, and synaptogenesis (Yong, 2005). In the adult CNS there is a decrease in MMPs levels, and MMPs expression alters mainly in response to neuronal activity. In response to injury and neurological diseases, the activity of MMPs is upregulated, with different MMPs characterized by specific temporal profiles, indicating their involvement in the pathogenesis of distinct neurological conditions (Zhang et al., 2010). The most studied and abundant types of MMPs in the rodent CNS are MMP-2 and MMP-9, which are immunolocalized in neurons, astrocytes, endothelial cells, and myelinated fibers (Noble et al., 2002; Planas et al., 2001; Rosenberg, 2001), but other MMPs have been found as well (Wells et al., 2003). MMPs have been detected also in the human brain with, for example, astrocytes expressing both MMP-2 and MMP-9, microglia expressing MMP-2, MMP-9 and MMP-7, and neurons expressing MMP-1 (Buss et al., 2007; Cossins et al., 1997; Cuzner et al., 1996; Rosenberg, 2001).
MMPs have been implicated in the regulation of different aspects of neural plasticity, including synapse remodeling, dendritic rearrangement, axon regeneration and neurogenesis, through interaction and modification of the ECM (Fujioka et al., 2012). The mature ECM environment exerts mainly an inhibitory action on the structural plasticity of neuronal networks. This inhibitory action may be counteracted by proteolytic alteration of the ECM components enabling a reactivation of plasticity and remodeling of neural circuits in the adult CNS in response to internal or external stimuli (Dityatev et al., 2010). Indeed, it has been shown that, in response to LTP induction in hippocampal slices from rats, MMP-9 is activated rapidly at perisynaptic sites and is essential for the maintenance of LTP (Meighan et al., 2006; Nagy et al., 2006). Moreover, it has been reported that spine enlargement during hippocampal LTP is dependent upon MMP-9 activation (Wang et al., 2008). Together these data suggest that proteolytic activity of MMP-9 is critical for synaptic strengthening and dendritic remodeling. MMPs participate in the regulation of synaptic plasticity also in an indirect way by processing the pro-forms of some neurotrophic factors into mature forms, such as NGF and BDNF, which are activated by MMP-3 and MMP-7, and cytokines, such as TNF-α, which is activated by several MMPs (Lee et al., 2001; Sternlicht & Werb, 2001).
MMPs have multiple effects in the CNS after traumatic injury (Zhang et al., 2010). Some findings suggest that in the acute period MMPs may play a protective role by suppressing apoptotic signaling pathways after injury, e.g. through MMP-3-dependent cleavage of the Fas ligand (FasL) from the neuronal surface (Wetzel et al., 2003; Wetzel et al., 2004). In contrast, data from experimental models of SCI suggest detrimental effects during the early phases of injury. Murine models of SCI showed the upregulation of MMP-9 activity on day 1 post-injury, followed by a gradual decline to negligible levels by day 14, whereas MMP-2 activity began to rise later, reaching a peak between day 7 and 14 post-injury (de Castro et al., 2000; Goussev et al., 2003). Upregulation of MMPs at the site of injury is likely due to their overexpression in activated microglia, astrocytes, and infiltrated leukocytes. The latter exploit MMPs to facilitate their entry into the damaged site through disintegration of the BSCB, thereby amplifying the local inflammatory response (de Castro et al., 2000; Noble et al., 2002). Elevated local levels of MMPs are also thought to contribute to demyelination and necrosis of spinal cord cells during the phase of secondary injury (Zhang et al., 2010). While MMPs are harmful during the acute phase, there is growing evidence that they can also support recovery during the later phase of SCI by promoting remyelination and axon regeneration. The regenerative potential of injured axons is stimulated by the breakdown of ECM inhibitory proteins associated with the formation of the glial scar, thereby creating an environment to facilitate structural and functional recovery (Liu et al., 2007). Particularly relevant to the CNS glial scar is MMP-2, which is involved in the degradation of neurocan and versican chondroitin sulfate proteoglycans (CSPGs), and MMP-3, capable of degrading tenascin-C, brevican, neurocan, NG2, phosphacan, and versican CSPGs (Imai et al., 1994; Muir et al., 2002; Siri et al., 1995; Turk et al., 2001). The peak of post-injury expression of these CSPG components coincides with the activity peak of the corresponding MMPs, suggesting that MMPs are induced in scar tissue of injured adult spinal cord in order to permit axonal regrowth (Duchossoy et al., 2001; Jones et al., 2003). An indirect trophic role in axon regeneration and sprouting has also been hypothesized for MMPs through a process of release of previously ECM-sequestered growth factors and their transformation from unprocessed pro-forms into mature active forms.
Upregulation of MMPs has been detected also in humans after SCI, with similar expression profiles to those observed in animal studies. The serum levels of MMP-2, MMP-8, MMP-9, MMP-10 and MMP-12 were measured in a prospective explorative study over a 12-week period in 115 patients after acute SCI (Moghaddam et al., 2017). To evaluate the impact of the MMPs expression profile on neurological outcome after traumatic SCI, 10 patients with neurological remission were selected and compared to 10 patients without remission, according to ASIA score. While MMP-10 and MMP-12 had a low serum level and were excluded from further analyses, MMP-2, MMP-8 and MMP-9 showed distinctive expression patterns between the two experimental groups. Mean MMP-2 values were mostly higher in patients without neurological improvement during the first week, with both groups peaking at 24 hours after SCI followed by a gradual decrease to plateau at day 7, when the expression curve of the two groups overlapped. Although no significant differences were found at any time, these data are in line with findings from animal studies suggesting that MMP-2 may be involved in functional recovery after SCI through regulation of glial scar formation and axonal plasticity (Hsu et al., 2006). Since these processes are particularly active in the first 7 days post-injury, it has been estimated that this period represents the time span of tissue remodeling (Yokobori et al., 2015). Mean MMP-8 values were higher in the group without remission during almost the entire period of follow-up, characterized by a significant difference between groups at admission and 24 h after SCI. The peak was reached at 7 days in both groups, followed by a decrease with return to their initial level and stabilization at day 14. MMP-9 levels were higher in the group without remission at admission but converged with those of the remission group within the first 4 h, thereafter proceeding with a similar pattern until 14 days after SCI. The group with neurological improvement showed significantly decreased levels 1 month after and remained at the same level for the rest of the evaluation, while the group without remission maintained the same values from 14 days to 2 months after trauma and, then, decreased. Based on these data a prognostic model was created which allowed to differentiate between neurological remission and lack of neurological remission in 97%of cases. The predictive value of this model is an important finding as it indicates that MMPs have good potential as a biomarker for stratifying injury severity and predicting neurological outcome, though it should be interpreted with caution due to the small sample size of the study.
MicroRNAs
MicroRNAs (miRNAs) are a class of small non-coding single-stranded RNAs composed of 21–24 nucleotides that play an important role in post-transcriptional regulation of gene expression. MiRNAs silence the expression of genes via a mechanism of base-pairing with complementary sequences in the target mRNA leading to suppression of translation, cleavage or degradation of the target mRNA, and thus influence the final protein levels (Guo et al., 2010; Hydbring & Badalian-Very, 2013; Lim et al., 2005). A single miRNA is able to simultaneously regulate the expression of many genes since a perfect match with a sequence of the target mRNA is not required (Hydbring & Badalian-Very, 2013). Considering the huge number of possible targets, the final effect of a single miRNA is not unique, but rather depends on the interaction with other miRNAs and their spatial and temporal expression patterns. Through the modulation of about 60%of all genes in the human genome, miRNAs regulate numerous biological processes in health and disease (Friedman et al., 2009; Hydbring & Badalian-Very, 2013). In line with their function, most miRNAs are located intracellularly; however some of them, commonly known as circulating miRNAs, can also be found in the extracellular environment due to their small size and high stability (Kim, 2015). Since they show a high stability also in human serum, circulating miRNAs have been vastly investigated as potential biomarkers to define the state of disease and the prognosis. Dysregulation of miRNAs expression has been strongly linked to the pathophysiology of malignancy where miRNAs play a dual role, promoting both oncogenesis and tumor-suppression in a dynamic process of carcinogenesis (Malumbres, 2013). Besides cancer, various other pathological conditions including numerous cardiovascular and neurological diseases show altered expression levels of miRNAs (Rao et al., 2013; Reid et al., 2011).
A wide spectrum of miRNAs has been identified in the mammalian CNS where they play an important role in cell differentiation, organogenesis and dendrite morphogenesis during neurodevelopment (Kosik, 2006). In the adult brain, miRNAs are involved in the regulation of neural plasticity mechanisms (Bredy et al., 2011; Gao et al., 2010). Dendritic structure and function of mature neurons are controlled in an activity-dependent manner by mRNAs expressed and translated locally at dendritic sites, enhancing in this fashion the temporal and spatial computational capacity of the CNS (Martin & Zukin, 2006; Wang et al., 2010). Well-studied examples are miR-132 and miR-134, both modulated by neuronal activity and necessary for activity-dependent synaptic protein synthesis, remodeling and plasticity (Ye et al., 2016). A growing body of additional evidence suggests that the structural and functional nature of miRNAs is perfectly suited to their assuming rapid control of molecular cascades at the synaptic sites in response to extracellular signals; miRNAs thus play a powerful role in various aspects of neural circuits remodeling (Ye et al., 2016).
The issue of a time-dependent expression of miRNAs after SCI has been repeatedly addressed (Rodrigues et al., 2018). It has been shown that about 300 miRNAs are aberrantly expressed after SCI in rats, with a significant overexpression of 60 miRNAs and a reduction of 37 miRNAs, suggesting their potential role in the regulation of a number of secondary injury responses including inflammation, apoptosis, oxidative stress, and remodeling of neural networks (Bakhit et al., 1991; Jin et al., 2014; Ning et al., 2014). In the acute phase, on days 1, 3, and 7 after the injury, an altered expression pattern was found at the site of lesion with a few miRNAs being upregulated, such as miR-223, miR-21, miR-219-5p, and miR-146a, and most being downregulated, e.g. miR-107 and miR-29c that were significantly repressed at day 3. Some miRNAs with an altered expression profile have been associated with modulation of cell death through different pathways, indicating an important effect on apoptosis after SCI (Yunta et al., 2012). Involvement of miRNAs in post-injury circuit remodeling has been studied in a rat model of SCI subjected to a specific exercise program as a paradigm to promote spinal cord plasticity (Liu et al., 2012). In this study, variations of miR-21 and miR-199-3p expression in spinal cord tissue were found in association with altered levels of phosphatase and tensin homolog (PTEN) mRNA and mechanistic target of rapamycin (mTOR) mRNA, two proteins shown to be involved in the regeneration and plasticity of injured spinal cord (Park et al., 2008). This study provides evidence that a behavioral paradigm can alter miRNA levels in order to modulate neural plasticity gene expression (Liu et al., 2012). It has been shown that expression of other genes involved in neural remodeling after injury, such as BDNF and the cell division cycle 42 gene, CDC42, is regulated by a spectrum of miRNAs (Liu et al., 2016). BDNF can be influenced by miR-183, miR-195, miR-30a, miR-182, miR-381, miR-300-3p, and miR-325-5p, while CDC42 can be influenced by miR-185, miR-329, miR-340-5p, miR-381, and miR-383. Several miRNAs are thought to promote these two genes that play a critical role in self-repair after injury (Liu et al., 2016).
Driven by the growing evidence of their altered expression profile and involvement in multiple critical SCI processes, miRNAs have already been tested as a SCI prognostic biomarker. One of the first studies identified miR-9, miR-84-5p and miR-219 as potential prognostic biomarkers for SCI severity and functional outcome in mice since, in the acute phase, the serum levels of all three miRNAs were upregulated relative to the severity of SCI within 12 h after injury (Hachisuka et al., 2014). Another work analyzed the expression of a wider spectrum of miRNAs (more than 300) in a porcine model of thoracic SCI on day 1, 3, and 5 after SCI (Tigchelaar et al., 2017). It found that 58, 21, 9, and 7 miRNAs were significantly upregulated after severe, moderate, and mild SCI and sham surgery, respectively. A strong correlation was found between total miRNAs expression at day 1 and 3 post-injury and neurological outcome at 12 weeks post injury, suggesting that miRNAs may be a promising predictor of functional recovery in the early stages of injury. This study is interesting because it identifies an alteration of the global quantity of miRNAs as a prognostic factor, rather than focusing on specific miRNAs, with the greatest changes in miRNAs number and magnitude correlating with injury severity. As a rationale, the authors hypothesized that massive cell death occurring within damaged spinal cord tissue leads to the release of the cellular content in the circulation, thereby resulting in reduction in miRNAs levels at the injury site, as observed in the above-cited studies (Yunta et al., 2012), and enhancement of global serum miRNAs levels (Tigchelaar et al., 2017).
Evidence from animal pain models indicate that miRNAs regulate nociception at multiple levels, including dorsal root ganglia, the spinal dorsal horn and the brain, through the post-transcriptional modulation of genes implicated in pain generation and maintenance (Andersen et al., 2014). In line with these findings, altered miRNAs expression has been associated also with neuropathic and inflammatory pain following SCI. In a rat model of compressive SCI, the expression of miR-218 was significantly upregulated at the site of lesion relative to that of the sham group on post-operative day 1, 3, 7 and 14, in association with neuropathic pain symptoms. When miR-218 was downregulated in rats, pain behavior and inflammation decreased, suggesting that miR-218 indeed modulates pain perception, with the JAK/STAT3 signaling cascade being identified as the main molecular pathway to be affected (Li & Zhao, 2016). Besides the alteration of miRNAs at the site of injury, aberrant expression has been found also at the distal segments of the spinal cord with respect to the injury site (Strickland et al., 2014). Persistent altered expression of miR-1, miR-124, and miR-129-2 at the injury site, measured at days 2 and 15 post-injury, predicted the nociceptive response mediated by the spinal regions distal to the site of injury, suggesting a molecular mechanism for the interaction of SCI with adaptation of functionally intact distal sensorimotor circuitry (Strickland et al., 2014). While there is a growing body of evidence supporting the clinical application of miRNAs as a valid indicator of neurological outcome, onset and severity of complications and treatment response, further clinical studies are necessary to identify the importance of each subtype of miRNAs for the management of different aspects of SCI.
Discussion
In the above section, we have discussed the temporal dynamics of potential humoral SCI biomarkers involved in a wide spectrum of neural plasticity mechanisms (see Tables 1 2). Even though proof-of-principle evidence has emerged from animal studies, and initial data from clinical investigations support the trends observed in experimental models, the nature and timing of the human lesion are substantially different from those of animal models of SCI. Hence, the pattern of temporal expression of plasticity-related biomarkers in patients must be more precisely identified before it can be of clinical relevance.
Available data suggest a dual role for some of the proposed biomarkers based on their level of concentration and the time-window of SCI. As indicated above, alterations of BDNF, TNF-α and some MMPs (such as MMP-9) have been associated with poor outcome during the acute phase, while in the chronic phase they are factors that promote functional recovery. These findings suggest that, when considering indicators of neural plasticity in the context of SCI, the expression pattern of biomarkers needs to be evaluated over time, rather than measured at a single time point. The need for a global point of view is even more evident in the case of miRNAs, where a clear correlation has been shown between the total number of altered miRNAs and severity of SCI. In addition to predicting functional recovery, plasticity-related biomarkers have repeatedly shown to be involved in the pathophysiology of clinical complications of SCI. For example, BDNF, NGF, TNF-α and miRNAs are all associated with the development of neuropathic pain. It could be interesting to evaluate whether monitoring their concentrations at different time-points may be a useful screening tool to detect and treat this frequent and debilitating complication in the early phase. The possible contribution of peripheral sources to total serum biomarker concentration should also be taken into account in some cases, e.g. with TNF-α, in order to evaluate to what extent systemic factors influence the onset of neuropathic pain and whether their treatment may prevent or ameliorate this complication. IGF-1 is another biomarker secreted in physiological conditions both locally in the CNS and peripherally. So far, it has not been associated with maladaptive plasticity, but rather seems to be involved in mechanisms promoting functional recovery after SCI. The circulating levels of IGF-1 increase gradually starting from the subacute phase, as a possible local and systemic response to neural tissue damage (Logan et al., 1994), and are at higher levels during all evaluated time-points in the group with better neurological outcome. This is not surprising considering the multiple evidence of IGF-1’s pleiotropic effect on neural plasticity (Dyer et al., 2016) and on post-injury neural protection, axonal regeneration and astrocytic gliosis in scar formation, particularly evident in TBI (Mangiola et al., 2015), a pathological condition with similar pathophysiological mechanisms (Kwon et al., 2019). It is important to note that IGF-1 acts in synergy with other neurotrophic factors and this interplay is well established for BDNF (Mattson et al., 2004), which acts as a downstream effector in the molecular cascade promoting activity dependent neural plasticity driven by physical exercise and environmental enrichment strategies (Baroncelli et al., 2010; Sale et al., 2014).
A dual role should also be considered for neurotrophins as a subgroup of SCI biomarkers since, as has been vastly demonstrated (see above), the biological actions of neurotrophin precursors (e.g. mediating apoptotic cell death) are in net contrast to those of the mature forms involved in neural protection and plasticity. Therefore, the conversion of the pro-forms to the mature forms appears to be a critical step for the final balance and their prevalent biological action. An increased transformation into mature neurotrophins has been associated with activity dependent neural plasticity and may be favored by physical activity and exercise, as demonstrated for BDNF (Berchtold et al., 2002; Cotman & Berchtold, 2002). In pathological conditions, this process seems to be altered in the acute phase, leading to the upregulation of the pro-forms and subsequent enhancement of the mature forms, through increased expression of proteolytic enzymes, such as MMP-9 and MMP-2 (Planas et al., 2001; Rosell et al., 2006). This has been shown for BDNF signaling in a model of ischemic stroke (Rahman et al., 2018) and was interpreted as a possible reactive attempt of the perilesional tissue to counteract the damage. While MMPs in this context play a beneficial role, they can also mediate deleterious effects on both the blood-brain barrier (BBB) and the BSCB, increasing their permeability and contributing to additional tissue loss. As yet, we have limited data on the fine regulation of the conversion of the immature forms of neurotrophins to the mature products in the specific case of SCI. Therefore, it would be important to evaluate the temporal profile of the ratio immature/mature neurotrophins in future studies to better understand the contribution of the activated pathways on functional outcomes.
Together with neural and glial tissue damage and functional impairment, dysfunction of the BSCB due to both direct mechanical insult and neuroinflammation is an equally important element contributing to the progression of SCI. While it is clear that BSCB disruption occurs within the first few minutes after mechanical insult to the spinal cord, findings regarding the timing of restoration of its function are not uniform and differ depending on the techniques applied (Bartanusz et al., 2011). However, data from animal studies suggest that the BSCB permeability gradually decreases over time. Its dysfunction was reported to persist as long as 56 days after injury (Cohen et al., 2009). Since the integrity of the BSCB is undoubtedly important for the structural and functional outcome after SCI and its gradual restoration over time is positively correlated with motor improvement in rats (Cohen et al., 2009), the BSCB should also be taken into careful consideration when studying the dynamics of serum biomarkers. When the BSCB is disrupted, the proteins deriving from the damaged spinal cord parenchyma may cross the barrier and move into the blood circulation. An example is neural and glial structural proteins, such as the family of neurofilaments and glial fibrillary acid protein (GFAP), which have been vastly studied as early prognostic serum biomarkers in both animal and clinical studies (Albayar et al., 2019). However, without information about the integrity of the BSCB, it may be difficult to discriminate to what extent the variations of a serum biomarker reflect the ongoing damaging or healing process at the site of injury or whether they are influenced by the BSCB’s permeability. The interpretation is even more complex in the case of plasticity-related biomarkers since many of them are not exclusively secreted in the spinal cord. Increased BSCB permeability also enables the increased influx of peripherally secreted biomarkers from the circulation to the lesion site. Therefore, information about the modifications of BSCB permeability during the post-injury period may help better understand to what extent peripherally secreted biomarkers, involved in the regulation of plasticity, may indeed cross the BSCB and contribute to the healing process. Functional integrity of the BSCB in the clinical setting can be assessed by imaging techniques, in particular by T1-weighted dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) (Heye et al., 2014). While DCE-MRI has been shown to be a sensitive and noninvasive technique to study the BSCB, it is costly and unsuitable for serial applications. Therefore, the concept of identifying serum molecular biomarkers as an alternative to evaluate BBB disruption in different pathological conditions (Kadry et al., 2020) represents a good solution for studying BSCB dysfunction in SCI. As proposed in a review on BBB biomarkers in ischemic stroke (Li et al., 2018), possible candidates for peripheral biomarkers may be structural elements of the BSCB released in the circulation following damage, such as occludin, cellular fibronectin and microvascular endothelial cells or other neural and glial proteins that are abundant in the spinal cord and can easily cross the injured membrane. These include, for example, S100 calcium-binding protein B or ubiquitin carboxyl-terminal hydrolase isozyme. Since MMPs are involved in the degradation of structural components of the BSCB, they could also be investigated as possible indicators of the barrier permeability. MMP-9 may be of particular relevance for studying BSCB disruption since it is involved in the degradation of the basement membrane. Moreover, it has been shown in clinical studies that peripheral levels of MMP-9 are independently associated with BBB disruption (Castellanos et al., 2007).
Conclusions
In this review, we have focused on potential humoral biomarkers involved in the functional and structural plasticity occurring after traumatic SCI and show that there is a complex interplay of multiple factors. We analyzed data from both preclinical studies, based on experimental models of SCI, and clinical trials appraising possible candidate biomarkers with a well-established role in regulating plasticity mechanisms. Since the basic proof-of-principle evidence in the field is still based on data from animal models, it is necessary to evaluate the feasibility and reliability of the proposed biomarkers in a clinical setting. Considering the complexity and heterogeneity of traumatic SCI, it is not realistic to expect information coming from biomarkers of neuroplasticity to be exhaustive in terms of prognosis for functional recovery. In line with Thelin et al. (2019), we argue that only a combination of biomarkers involved in different pathophysiological processes (see Fig. 1 for a summary), including structural biomarkers originating from neural and glial cells, biomarkers of neuroinflammation, biomarkers of neural plasticity and finally BSCB permeability biomarkers, can refine the prediction models of functional outcome in SCI.
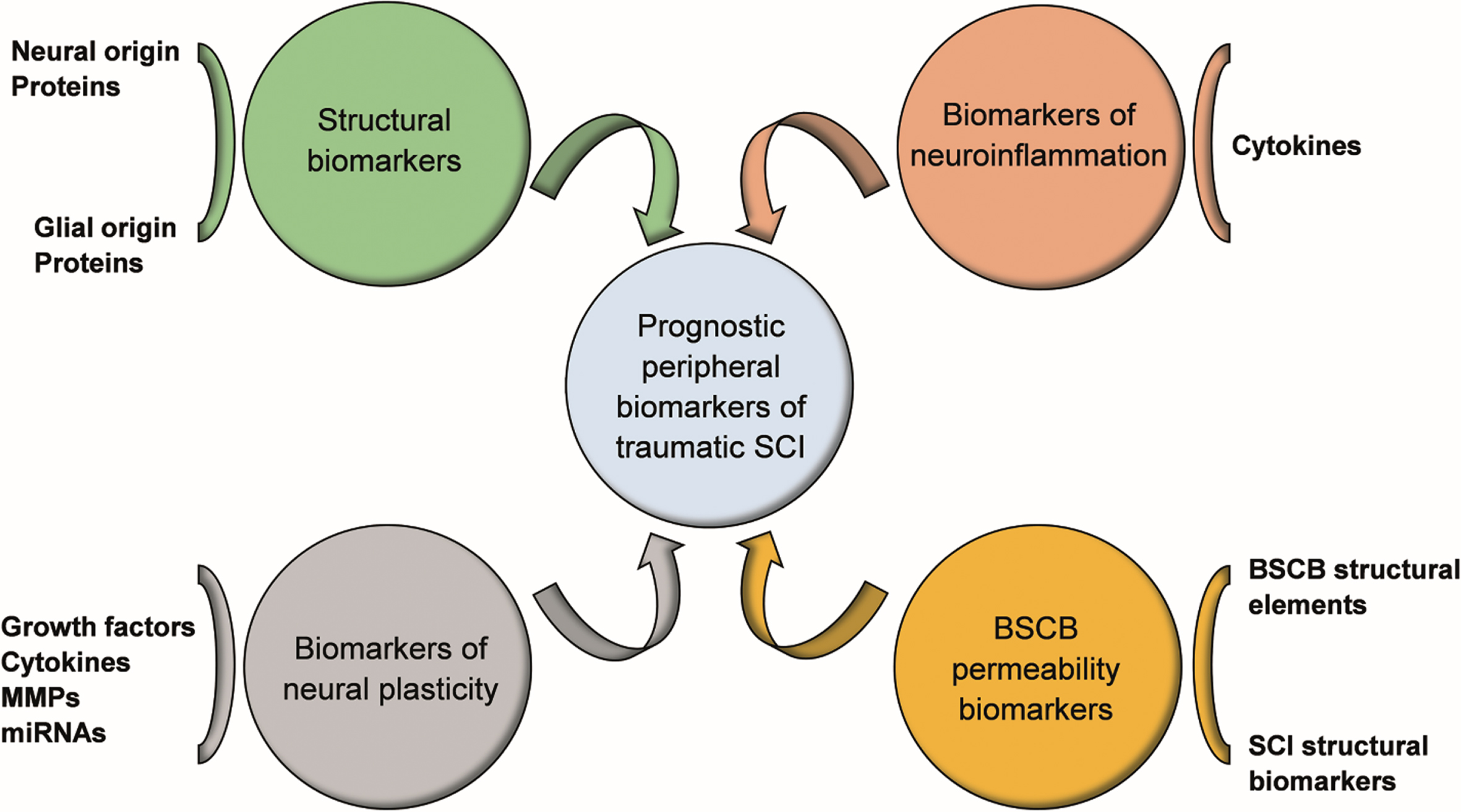
Future perspectives for humoral SCI biomarkers. Combination of biomarkers reflecting different pathophysiological processes of SCI, including structural biomarkers originating from neural and glial cells, biomarkers of neuroinflammation, biomarkers of neural plasticity and BSCB permeability biomarkers, may improve the prognosis of functional outcome and guide the selection of appropriate therapeutic strategies. Abbreviations: SCI, spinal cord injury; BSCB, blood spinal cord barrier; MMPs, Matrix Metalloproteinases; miRNAs, microRNAs.
Conflicts of interest
The authors have no conflict of interest to declare.
Authors’ contribution
Tatjana Begenisic and Daniela Rossi contributed to the conception and design of the work. Tatjana Begenisic drafted the work and edited the manuscript, tables and figures. Chiara Pavese and Beatrice Aiachini provided a final revision of the abstract and article. Daniela Rossi and Antonio Nardone contributed to editing the manuscript and figures and tables and provided a final revision of the abstract and article. All authors approved the final draft.
Footnotes
Acknowledgments
This research was partly supported by the Ricerca Corrente funding scheme of the Ministry of Health, Italy. It did not receive any specific grant from funding agencies in the commercial or for-profit sectors.