
Abstract
BACKGROUND:
Liver fibrosis is a progressive liver disease with increasing incidence, yet its underlying pathogenic mechanisms remain incompletely understood.
OBJECTIVE
: This study aims to explore potential therapeutic targets for liver fibrosis using weighted gene co-expression network analysis (WGCNA) and experimental validation.
METHODS:
We retrieved the microarray data (GSE174099) from the GEO database and performed differential expression analysis and WGCNA to identify co-expression modules associated with liver fibrosis. A module with the highest correlation to liver fibrosis was selected for further analysis. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were conducted to investigate the biological functions and signaling pathways of the identified genes. Protein-protein interaction (PPI) networks were constructed using the STRING database. The correlation between core genes and immune cells was analyzed with the CIBERSORT algorithm. Additionally, pathological and molecular biology experiments were performed to validate the expression levels of core genes in liver tissue, including HE and Masson staining, immunohistochemistry, RT-qPCR, and Western blotting.
RESULTS:
We identified a total of 86 intersecting genes from the differential expression analysis and WGCNA. GO enrichment analysis revealed that these genes were involved in processes such as cellular response to cAMP, collagen-containing extracellular matrix, and G protein-coupled receptor binding. KEGG pathway analysis highlighted the involvement of these genes in pathways like Cell Adhesion Molecules and the PI3K-Akt signaling pathway. Using Cytoscape software, we identified four core genes: Cftr, Cldn4, Map2, and Spp1. Pathological examinations showed that the experimental group exhibited significant fibrous tissue proliferation compared to the control group. Immunohistochemistry, RT-qPCR, and Western blotting analyses confirmed that these core genes were significantly upregulated in the experimental group (
CONCLUSION:
This study identified four key genes (Cftr, Cldn4, Map2, Spp1) that are significantly associated with liver fibrosis. These genes are upregulated in liver fibrosis and could potentially as biomarkers for diagnosis and targets for therapeutic interventions.
Introduction
Liver fibrosis is a prevalent hepatic disorder characterized by the pathological accumulation of extracellular matrix (ECM) in response to chronic liver injury caused by various factors, such as intoxication, viral infections, autoimmune diseases, and metabolic or genetic disorders. The global prevalence of liver fibrosis is estimated to range from 4.5% to 9%, resulting in approximately one million deaths annually and ranking as the 11th leading cause of mortality worldwide [1, 2] The persistent presence of liver fibrosis often progresses to cirrhosis, resulting in irreversible structural damage to the liver and an increased susceptibility to hepatocellular carcinoma, thereby significantly elevating mortality risk among patients with hepatic ailments [3, 4]. Despite significant advancements in the diagnosis and treatment of liver fibrosis in recent years [5], therapeutic outcomes remain unsatisfactory, as there is currently no approved drug available for the treatment of liver fibrosis [6]. The primary approach to treatment involves addressing or controlling the underlying causes; however, this alone proves insufficient in preventing the progression from liver fibrosis to cirrhosis [7]. Given the continuous rise in affected individuals, it is imperative to further elucidate the molecular pathogenesis of this disease to facilitate early intervention and novel drug development, thereby delaying advanced liver fibrosis.
Bioinformatics technology and high-throughput sequencing techniques, along with the establishment and enhancement of disease databases, have rapidly advanced, providing a theoretical foundation to elucidate the pathogenesis of liver fibrosis and identify novel therapeutic targets [8]. However, conventional bioinformatics methods may fail to recognize genes that play crucial roles in liver fibrosis pathogenesis but exhibit minimal differences at the gene expression level. Because differentially expressed genes may not fully capture the intricacies of liver fibrosis, we employed a systematic analysis method known as WGCNA to identify pivotal genes and signaling pathways implicated in the pathogenesis of liver fibrosis. WGCNA, a comprehensive biological approach, primarily dissects gene modules associated with specific phenotypic features across multiple samples, unraveling intricate interactions and regulatory networks among genes to gain profound insights into gene regulation mechanisms. Researchers have extensively applied this methodology to investigate the pathogenesis of diverse diseases, including liver fibrosis [9, 10].
In this study, we conducted an analysis of microarray data from liver fibrosis and normal liver tissue to identify differentially expressed genes (DEGs). Subsequently, utilizing the WGCNA method, we identified modules that exhibited the highest correlation with liver fibrosis, significantly narrowing down the pool of candidate genes. Furthermore, we employed other comprehensive analytical approaches to ascertain potential key genes and evaluate the extent of immune infiltration between liver fibrosis and normal liver tissue. In conclusion, our findings reveal four key genes, Cftr, Cldn4, Map2, and Spp1, which are considered promising diagnostic biomarkers that play crucial roles in the initiation and progression of liver fibrosis.These discoveries hold significant implications for unraveling its pathogenesis and developing effective treatment strategies.
Materials and methods
Data collection and preprocessing
The gene expression profiles related to liver fibrosis analyzed in this study were obtained from the Gene Expression Omnibus database, accessible at
Analysis of differentially expressed genes
The downloaded expression data matrix was normalized using the R software. Differential expression analysis was performed on the normalized expression profiles using the “limma” package, and genes with differential expression were filtered using
Weighted gene co-expression network analysis
The R package “WGCNA” was used to generate the sampleTree in order to identify any potential outliers. Subsequently, the weighted gene co-expression network of the liver fibrosis expression matrix was created using a one-step approach. The adjacency matrix was converted into a topological overlap matrix (TOM), with a scale free
Acquisition of core genes and analysis of gene enrichment
The study utilized Bioinformatics and Evolutionary Genomics methods to intersect the outcomes of differential expression analysis and WGCNA. Additionally, Venn diagrams were constructed to identify the common set of genes associated with the progression of liver fibrosis. The core genes were subjected to gene ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis using the R package "clusterProfiler". The utilization of GO enrichment analysis was employed to acquire the enrichment outcomes of fundamental genes in Molecular Function (MF), Cellular Components (CC), and Biological Process (BP). The enrichment results of key gene signaling pathways were obtained through the use of KEGG enrichment analysis, using screening criteria of
Construction of protein–protein interaction (PPI) networks of core genes
The protein-protein interaction (PPI) network of genes related to liver fibrosis was constructed using the STRING database (
Analysis of immunological associations
The CIBERSORT analysis was employed to elucidate the infiltration levels of 22 immune cells in rats with liver fibrosis. Additionally, correlation analysis was conducted to establish the relationship between core genes and immune cell infiltration in the context of liver fibrosis.
The establishment of a rat liver fibrosis model and the collecting of specimens
Acquisition of experimental animals
A total of six male Sprague-Dawley rats, aged 6–7 weeks and weighing between 180–200 g, were acquired and thereafter kept in the animal facility with a specific pathogen-free (SPF) designation at the Experimental Animal Center of Kunming Medical University. The experimental procedure adhered to ethical guidelines and underwent scrutiny and approval by the Ethics Committee of Calmette Hospital, affiliated with Kunming Medical University.
Establishment of animal models and procurement of specimens
The rats were divided into two groups through the utilization of a random number table method. The control group consisted of three rats, whereas the experimental group, specifically the liver fibrosis group, also comprised three rats. Prior to surgery, all rats were fasted for a duration of 12 hours. The rats in the control group were subjected to a simulated procedure, which involved the administration of anesthesia, performing a laparotomy, dissociating the common bile duct, and afterwards closing the abdomen. The rats in the liver fibrosis group were subjected to anesthesia, underwent laparotomy, and the common bile duct was doubly ligated with 3-0 silk suture. The initial ligation occurred near the hepatic duct junction, while the subsequent ligation was positioned above the entrance of the pancreatic duct. The common bile duct was cut between the ligatures, after which the abdominal cavity was sutured up. The rats were allowed to recover from anesthesia. Four weeks after surgery, the rats were euthanized, and liver and blood samples were collected for analysis of liver function, histology, immunohistochemistry, RNA, and protein.
Biochemical testing on serum
The serum levels of total bilirubin (TBIL), direct bilirubin (DBIL), indirect bilirubin (IBIL), alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP) were determined using a fully automated biochemical analyzer (ADVIA Chemistry XPT, SIEMENS, USA).
Histopathology examination
Liver tissue specimens from each group were obtained and afterwards immersed in a 4% paraformaldehyde solution (Bio sharp Life sciences, China) for a duration of 24 hours to ensure proper fixation. The fixed specimens were routinely dehydrated, paraffin-embedded, sectioned, and deparaffinized for histological and immunohistochemical analysis. The thickness of the paraffin sections was 4–5
The evaluation of liver fibrosis stage. The assessment of liver fibrosis grading was conducted utilizing the Metavir scoring system, which employs a 5-point scale to evaluate the severity of liver fibrosis. The scoring system includes the following categories: F0 indicating the absence of liver fibrosis, F1 indicating the absence of fibrous septa in the portal area, F2 indicating the presence of a limited number of fibrous septa in the portal area, F3 indicating structural disorganization of the septal fibrosis, and F4 indicating the presence of cirrhosis [11].
Immunohistochemical analysis
Liver tissue samples selected for immunohistochemistry staining in order to confirm the variations in expression between non-liver fibrosis and liver fibrosis tissues. The specimens were treated overnight at 4∘C with antibodies Cftr (1:100, 40734, Signalway Antibody, China), Cldn4 (1:100, ER63025, HUABIO, China), Map2 (1:200, ha500177, HUABIO, China), and Spp1 (1:100, 22952-1-ap, Proteintech, USA). Subsequently, they were incubated with a secondary antibody, specifically goat anti-rabbit IgG (1:500; G1213-100UL, Servicebio, China), and mixed for a duration of 30 minutes at 37∘C. Following this, the specimens underwent diamino-benzidine (DAB) staining. The sections were examined and imaged using a microscope (Olympus, Japan) to assess the degree of immunostaining. The resulting images were then analyzed using Image pro plus 6.0 software.
Real-Time quantitative polymerase chain reaction (RT-qPCR) analysis
The extraction of total RNA from rat liver tissue was performed using Trizol (Invitrogen, USA), followed by the synthesis of complementary single-stranded DNA (cDNA) (Servicebio, China) using reverse transcription. The quantification of cDNA targets was conducted using a CFX96TMReal-Time Fluorescent Quantitative PCR Detection System (Bio-Rad, USA). By conducting a thorough search in the NCBI database, we successfully retrieved the complete mRNA sequences of Cftr, Cldn4, Map2, Spp1, and GAPDH. Subsequently, highly specific and efficient primers were meticulously designed using Primer 5.0 software. These primers will be synthesized by Tsingke in China. The sequences are presented in Table 1. The SYBR Green premix (Servicebio, China) was utilized to duplicate each sample three times. The expression levels of the target genes were normalized to GAPDH, and the relative expression was determined using the 2 - ΔΔCt method.
Primers Used for RT-qPCR amplification
Primers Used for RT-qPCR amplification
RIPA lysis buffer (Servicebio, China) was used to prepare liver tissue. The BCA protein quantification reagent was used to ascertain the protein concentration. SDS-PAGE (10% gel) was used to separate total protein (40 mg/lane) from each sample, which was subsequently transferred onto PVDF membranes (EMD Millipore, USA).
Statistical analysis
SPSS 22.0 software was used for data analysis, and GraphPad Prism9.0 software was used for drawing. All quantitative data were presented as mean
Results
Differently expressed genes
The raw data underwent preprocessing. By applying the criteria of
Analysis of differential gene expression. (A) Heatmap illustrating the differential gene expression between normal liver tissue and liver fibrosis tissue. (B) Volcano plot of differentially expressed genes in normal liver tissue and liver fibrosis tissue sequencing data, with green indicating downregulated genes, red indicating upregulated genes, and black indicating genes with no significant difference. 
To identify key gene modules associated with the occurrence of liver fibrosis, we utilized WGCNA to analyze the relationship between all genes and the development of liver fibrosis in a liver fibrosis-related dataset (GSE174099) (Fig. 2A and B). Based on average linkage hierarchical clustering and soft thresholding power, seven gene modules were identified in the GSE174099 dataset(Fig. 2C).In the GSE174099 dataset, we observed a strong positive correlation between liver fibrosis and the brown module(
Reveal key modules associated with liver fibrosis in the GEO dataset through the utilization of WGCNA. (A) Analysis of scale independence and mean connectivity plots using WGCNA methodology. (B) The dendrogram of all genes in the GSE174099 dataset is hierarchically clustered using various measures (1-TOM). Each branch within the dendrogram represents an individual gene, while the color coding of each module signifies a co-expression module. (C) The GSE174099 dataset provides a heatmap illustrating the correlation between liver fibrosis and clustered gene modules, with each module presenting correlation coefficients and p-values. 
The intersection of genes exhibiting
Identification of a core gene set associated with liver fibrosis through comprehensive screening. (A) The Venn diagram depicts the overlap between differentially expressed genes in the GSE174099 dataset and genes affiliated with WGCNA modules exhibiting positive correlations. (B) Enrichment analysis of key genes associated with liver fibrosis using GO terms. (C) KEGG enrichment analysis was conducted on core genes associated with liver fibrosis. 
The investigation of the protein-protein interaction network of the key genes was conducted utilizing the STRING database and Cytoscape software (Fig. 4A). Based on this rationale, the four main genes, namely Cftr, Cldn4, Map2, and Spp1, were subjected to further screening using the degree plugin of Cytoscape program (see Fig. 4B).
Protein-Protein Interaction Network Associated with liver fibrosis.(A) The protein-protein interaction network of essential genes. (B) Protein interaction network diagram illustrating the core genes with high Degree scores (Note: The intensity of color corresponds to the ranking level). 
Using the CIBERSORT analysis method, we determined the infiltration levels of 22 immune cells in rats with liver fibrosis. Correlation analysis was performed to establish associations between core genes and immune cell infiltration levels in liver fibrosis, resulting in a heatmap (Fig. 5). The results demonstrate a positive correlation between Cftr expression and Plasma cells as well as Macrophages M1. Moreover, Cldn4 and Spp1 expression exhibit positive correlations with Mast cells resting. Additionally, Spp1 expression shows a negative correlation with Tregs and Dendritic cells activated.
Analysis of immune infiltration. A heatmap illustrating the correlation between the infiltration levels of 22 immune cells in liver fibrosis rats and core genes was generated. (Note: red squares represent positive correlations, blue squares indicate negative correlations, and darker color squares signify stronger correlations). 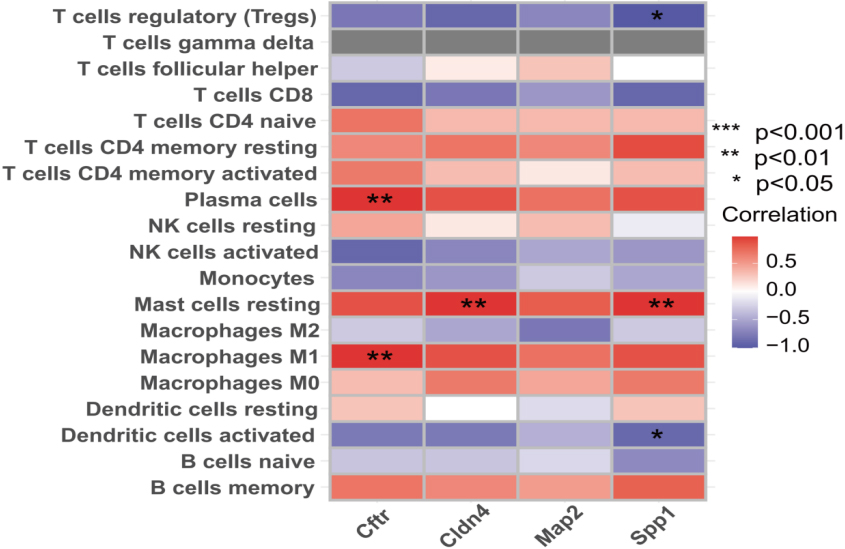
Compared to the control group, the liver fibrosis group exhibited significantly elevated levels of TBIL, DBIL, IBIL, ALT, AST and ALP (
(A) The levels of total bilirubin (TBIL), direct bilirubin (DBIL), indirect bilirubin (IBIL), alanine aminotransferase (ALT), aspartate aminotransferase (AST) and alkaline phosphatase (ALP) were compared between the control group and liver fibrosis group at 4 weeks post-surgery. (B) Morphological liver structure at 4 weeks post-surgery in the control and liver fibrosis groups. (C) Revealing the expression levels of Masson staining in both groups. (D) Evaluation of liver fibrosis using Metavir scores in two groups. (E) Evaluation of rat liver fibrosis model: Histological staining was performed on the livers of both control and liver fibrosis groups at 4 weeks post-surgery. HE-stained and Masson-stained micrographs were captured at magnifications of 100x and 200x, respectively. The data were presented as mean 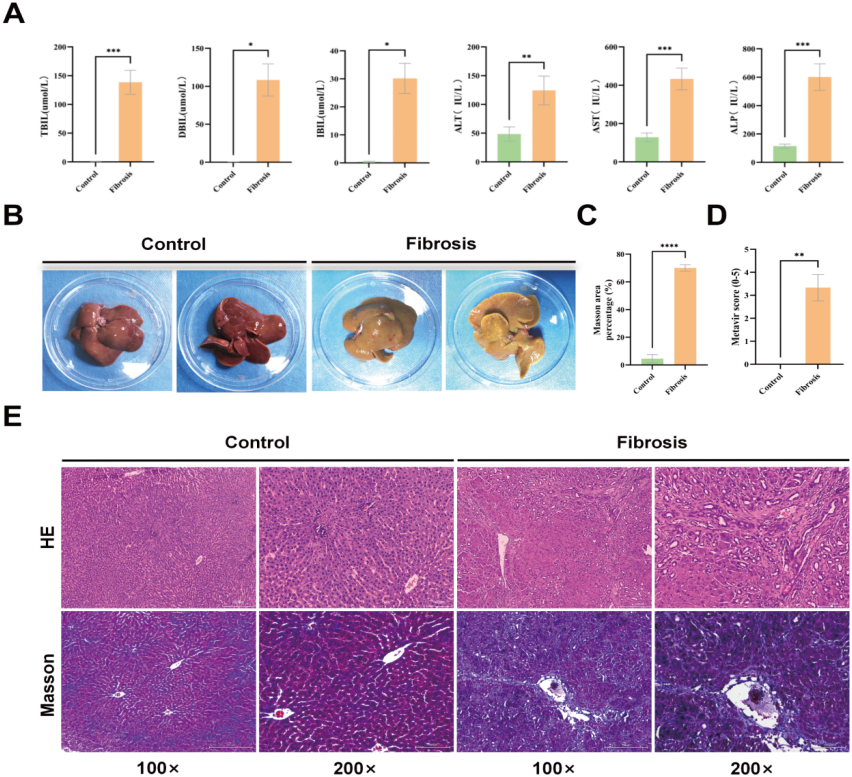
Morphological assessment of the liver
The construction of the rat liver fibrosis model was accomplished with success. In the control group, the livers of the rats exhibited a reddish-brown hue, a soft consistency, and a surface that was both smooth and characterised by sharp edges. The liver fibrosis group exhibited liver tissue in rats that displayed a yellowish hue, firm consistency, and a surface characterised by gritty granules and blunt edges (Fig. 6B).
Hematoxylin-eosin staining
Histological examination of liver tissues in each group was conducted using HE staining and the Metavir scoring system to evaluate the degree of liver fibrosis. Pathological examination of liver tissue sections in the control group showed that the hepatic lobule structure was intact, the liver cell cords were arranged radially around the central vein in an orderly manner, and no liver cell necrosis or lymphocyte infiltration was observed. The liver fibrosis score was determined to be F0 (Fig. 6E). Histopathological examination of the liver fibrosis group revealed aberrant proliferation of fibrous tissue within hepatic parenchyma, characterized by dense collagen fibers forming a fibrous septum. This was accompanied by disrupted lobular architecture, portal area enlargement, triangular bridging fibrosis formation, increased small bile duct proliferation, and diffuse lymphocytic infiltration. A statistically significant difference was observed between the liver fibrosis group and the control group post-surgery (
Masson staining
Masson’s staining of the control group showed a small amount of blue collagen fibers surrounding individual portal areas, with no observed fibrous tissue proliferation. However, in the liver fibrosis group, there was a significant presence of blue-stained fibrous tissue in the portal areas (Fig. 6E). Furthermore, there was a significant difference compared to the control group (
Expression of key genes in liver tissue
Immunohistochemical assessment of alterations in the protein expression levels of Cftr, Cldn4, Map2, and Spp1
In the control group liver tissue, hepatocytes and portal area exhibited a low expression of Cftr, Cldn4, Map2, and Spp1 proteins (Fig. 7A). However, in the liver fibrosis group, there was a significant upregulation of these proteins in both hepatocytes and portal area (Fig. 7A). The protein expression levels of Cftr, Cldn4, Map2, and Spp1 were significantly elevated in the liver fibrosis group compared to the control group (
(A) Micrographs of differentially expressed proteins, determined by immunohistochemistry, were captured in the livers of both control and liver fibrosis groups at 4 weeks postoperatively, with magnifications set at 100x and 200x respectively. (B) Immunohistochemistry was employed to assess the protein expression levels of Cftr, Cldn4, Map2, and Spp1 in both the control group and liver fibrosis group. (C) The relative expression levels of four genes in the control group and liver fibrosis group were quantified using RT-qPCR. (D) Validation of differentially expressed proteins in the control group and liver fibrosis group using Western blotting. (E) The protein expression levels of the control and liver fibrosis groups were validated through Western blotting. Results are presented as (mean 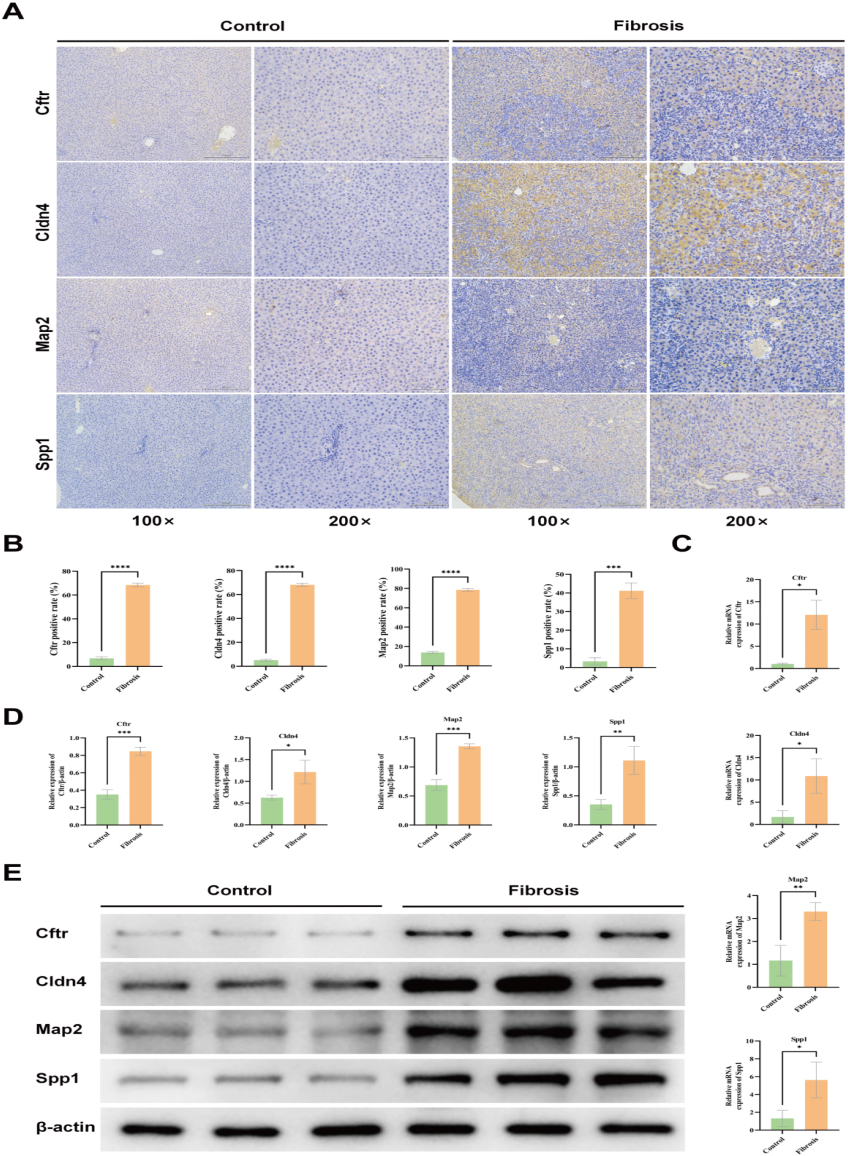
The reverse transcription-quantitative polymerase chain reaction (RT-qPCR) findings demonstrated that the predicted core genes exhibited increased expression in the context of liver fibrosis. Specifically, the mRNA expression levels of Cftr, Cldn4, Map2, and Spp1 were considerably elevated in the liver fibrosis group compared to the control group (
Protein levels of Cftr, Cldn4, Map2, and Spp1 were quantified by employing Western blotting analysis
The Western blotting assessment showed that the protein expression levels of Cftr, Cldn4, Map2, and Spp1 were reduced in the rat liver tissues of the control group. Conversely, in the liver fibrosis group, the expression levels of these proteins were notably elevated compared to the control group (
Discussion
The hallmark of liver fibrosis is the excessive accumulation of ECM components in the injured hepatic region. Drug, metabolic, infectious, or cholestatic insults typically induce chronic liver injury that leads to progressive fibrosis, which can progress to cirrhosis, hepatic failure, and hepatocellular carcinoma [12]. It represents a pivotal event in the pathogenesis and progression of chronic liver diseases. The malignant progression of liver fibrosis constitutes the primary cause of mortality on a global scale [13]. The pathogenesis of liver fibrosis is a complex, multi-step process that occurs over an extended period of time. However, the specific molecular mechanisms underlying its progression remain unclear, and effective treatment strategies are still lacking. As a result, it is critical to identify and validate key genes associated with liver fibrosis progression to gain insights into its pathogenesis and develop novel therapeutic approaches.
By employing bioinformatics methodologies and leveraging high-throughput sequencing data, the underlying mechanisms driving liver fibrosis progression can be comprehensively investigated, thereby providing robust evidence to support the development of therapeutic strategies targeting this pathological condition. To find genes that are differently expressed between the normal group and the liver fibrosis group in this study, we used differential analysis on the GSE174099 chip data that we got from the GEO database. After that, we used the WGCNA method to find seven co-expression modules and then chose highly correlated modules to get 86 genes that overlap with differentially expressed genes. We conducted GO and KEGG functional enrichment analyses on these highly correlated intersecting genes. Furthermore, through protein-protein interaction network analysis, we identified 86 potential hub genes, including Cftr, Cldn4, Map2, and Spp1 as key upregulated genes. Considering that GO and KEGG pathway analyses revealed their association primarily with inflammatory responses, gene enrichment analysis indicated immune cell infiltration in the liver fibrosis group. Furthermore, by examining liver histomorphology, testing biochemical functions, and conducting pathological examinations using HE and Masson staining on rats four weeks post-surgery, we have effectively created a model of rat liver fibrosis. We subjected the liver tissue samples from the experimental group to RT-qPCR, IHC, and Western blotting analyses in the present study. The obtained results revealed a significant up-regulation in the mRNA and protein expression levels of Cftr, Cldn4, Map2, and Spp1 when compared to the control group. These findings align with our initial predictions, thus validating our hypothesis. In summary, these technologies exhibit high sensitivity and specificity, facilitating the quantitative detection of gene and protein expression levels. They give complete details from transcription to translation, which makes sure that bioinformatics analysis results are correct and confirms the biological functions and regulatory roles of core genes in liver fibrosis.
The study used differential expression analysis to look into the biological functions and signaling pathways of 86 hub genes linked to the progression of liver fibrosis. This was done by using GO and KEGG enrichment analyses. The results demonstrated that these 86 hub genes are mainly enriched in cellular response to cAMP, collagen-containing extracellular matrix, and G protein-coupled receptor binding. In the process of liver fibrosis, the transformation of fibroblasts into myofibroblasts significantly enhances disease progression [14]. Cellular response to cAMP refers to the signaling and regulatory processes through which cells react to cyclic adenosine monophosphate (cAMP). As a second messenger, cAMP is produced by adenylate cyclase in response to activation of G protein-coupled receptors. It plays a pivotal role in regulating fibroblast function and has been demonstrated to promote regression of fibrosis, making it a potential target for decelerating the fibrotic process [15, 16]. Heightened cAMP levels can inhibit the activation of hepatic stellate cells (HSCs) and fibroblasts, curtail their proliferation, and lower their survival rate, all while impeding the synthesis of ECM proteins [15]. Numerous in vitro and in vivo experimental studies have demonstrated the pivotal role of multiple signaling pathways in the pathogenesis of liver fibrosis. In this study, KEGG pathway enrichment analysis revealed that differentially expressed genes were predominantly enriched in Cell adhesion molecules and the PI3K-Akt signaling pathway. Cell adhesion molecules play a pivotal role in liver fibrosis by regulating cell-cell and cell-ECM adhesion, as well as participating in the modulation of inflammatory responses [17]. In the context of liver fibrosis, cell adhesion molecules modulate adhesive interactions between hepatic cells and the ECM, influencing cellular responses to the matrix and promoting extensive release of Transforming growth factor beta (TGF-
In order to gain a deeper understanding of the pivotal genes that impact the advancement of liver fibrosis, this study utilized WGCNA and PPI methodologies alongside differential expression analysis. As a result, four crucially upregulated key genes were identified: Cftr, Cldn4, Map2, and Spp1. The Cftr gene, located on chromosome 7’s long arm, encodes the transmembrane conductance regulator protein known as Cystic Fibrosis Transmembrane Conductance Regulator (Cftr), which belongs to the ATP-binding cassette superfamily [22]. Cftr is widely distributed in epithelial cells across various tissues and plays a crucial role in activating cAMP-dependent chloride ion channels, thereby significantly contributing to cellular homeostasis and metabolism [23]. In the liver, the Cftr protein is prominently expressed on the apical membrane of bile duct cells, thereby influencing biliary transport and leading to the retention of toxic bile acids. Moreover, Cftr triggers the expression of the essential fibrotic factor MCP-1 in hepatocytes and bile duct cells, which, in conjunction with other factors, enhances hepatic stellate cell chemotaxis, culminating in peri-biliary fibrosis [24]. Previous research has demonstrated an escalating prevalence of Cftr gene mutations in patients diagnosed with primary sclerosing cholangitis (PSC). The aberration of Cftr is correlated with a higher incidence rate [25]. Moreover, findings from recent studies suggest that during the course of bile stasis, there is an upregulation of Cftr gene expression. It suggests that this alteration in gene expression might contribute to the pathogenesis of cholestatic liver diseases [26]. According to other research findings, the expression of the Cftr gene demonstrated a downward trend during the alleviation process of cholestasis-induced bile duct reactions and fibrosis in rats [27]. The findings of this research demonstrated a significant upregulation in the mRNA and protein expression levels of Cftr in the liver fibrosis group when compared to the control group. This outcome aligns with the current body of literature and potentially contributes to the pathogenesis and progression of liver fibrosis.
Claudin-4 (Cldn4) is an essential constituent of tight junctions (TJ), functioning as a transmembrane protein that plays a pivotal role in regulating paracellular permeability and maintaining the epithelial cell polarization state [28]. Cldn4 is frequently overexpressed in various malignant tumors, and its aberrant expression has been demonstrated to contribute to tumorigenesis [28]. Studies have revealed elevated levels of Cldn4 expression, specifically in hepatocellular carcinoma [29]. Importantly, it promotes epithelial-mesenchymal transition (EMT), thereby enhancing cell invasion and metastatic potential [30]. The upregulation of Cldn4 expression has been observed in both liver cancer and cirrhosis [31]. The findings indicated that the Cldn4 gene was observed to be expressed in hepatocytes situated in the vicinity of the regenerating nodules inside the cirrhotic liver. The expression of the Cldn4 gene in liver tissue affected by chronic hepatitis exhibits a positive correlation with the severity of fibrosis [32]. The results of our investigation demonstrate a significant upregulation in the expression level of the Cldn4 gene in liver fibrosis tissues compared to normal hepatic cells. Given that liver fibrosis represents an initial stage in the progression towards liver cirrhosis, these findings suggest that Cldn4 has potential as a diagnostic biomarker for detecting liver fibrosis.
Map2 (Microtubule-associated protein 2) is a pivotal cytoskeletal protein predominantly expressed in neurons, playing essential roles in microtubule nucleation and stabilization, facilitation of microtubule protein aggregation, organelle transport, and regulation of protein anchoring [33, 34]. The overexpression of Map2 protein has been observed in various tumors, and studies have demonstrated that its aberrant expression can enhance phosphorylation levels, diminish binding affinity with microtubules, and disrupt microtubule dynamics, leading to cellular cytoskeleton reorganization and structural abnormalities. Ultimately, this promotes the tumor’s invasive and metastatic capabilities [35]. In the research on liver fibrosis, the role of Map2 has not been fully explored. Recent studies have demonstrated the pivotal role of cytoskeletal reconstruction and dynamic microtubules in cellular remodeling and migration processes during liver fibrosis [36, 37, 38]. Our research revealed that during the progression of liver fibrosis, both mRNA and protein levels of Map2 were significantly elevated in the liver fibrosis group compared to the control group. Therefore, further investigation is warranted to explore the potential role of MAP2 in liver fibrosis.
Spp1 (secreted phosphoprotein 1, Spp1) plays diverse roles in the extracellular matrix by facilitating and regulating cellular-matrix interactions [39]. As an inflammatory cytokine, Spp1 demonstrates a range of activities associated with the development and progression of hepatitis, liver fibrosis, and liver cancer [40]. Studies have indicated that plasma levels of Spp1 can serve as an indicator for predicting liver fibrosis [41], and it is significantly correlated with the extent of fibrosis, hepatic dysfunction, portal hypertension, and hepatocellular carcinoma [42]. Spp1 plays a pivotal role in liver fibrosis by activating HSCs and inducing the deposition of collagen I through a transforming growth factor-beta (TGF-
GSEA enrichment analysis revealed the comprehensive immune profile of liver fibrosis. Our findings demonstrate a positive correlation between Cftr expression and plasma cells as well as macrophages M1. Conversely, Cldn4 and Spp1 expression exhibited a positive association with mast cells resting. Furthermore, Spp1 expression displayed a negative correlation with Tregs and activated dendritic cells. HSCs activation is a key step in liver fibrosis. Immune cells, especially macrophages, play a key regulatory role in HSCs activation [46]. M1 macrophages contribute to the inflammatory response by releasing pro-inflammatory cytokines, thereby exacerbating the progression of hepatic damage. Studies have demonstrated that in cases of chronic active hepatitis, M1 macrophages persist at elevated levels over extended periods, thereby perpetuating hepatic inflammation. The prolonged presence of M1 macrophages hinders the resolution of inflammation, initiates fibrosis, and facilitates scar formation [47]. Previous studies have reported an increased presence of plasma cells in liver fibrosis tissue; however, it is currently acknowledged that B lymphocytes contribute to and sustain liver fibrosis by regulating inflammation and restraining hepatic stellate cell aging [47]. Mast cells resting play a crucial role in tumor immunity, exhibiting significantly higher infiltration density in hepatocellular carcinoma and intrahepatic cholangiocarcinoma compared to normal liver tissue [48]. The heightened infiltration density of mast cells has been associated with an unfavorable prognosis among patients with hepatocellular carcinoma [49]. Regulatory T cells (Tregs) play a critical role in modulating the immune microenvironment and maintaining immune homeostasis [47]. Previous studies have demonstrated that a substantial population of highly activated and differentiated Tregs specifically localizes to the infiltrated liver in chronic HCV infection, effectively limiting the progression of fibrosis [50]. This finding suggests that an adequate presence of Tregs can impede the advancement of fibrotic processes by exerting negative regulation on the inflammatory response responsible for excessive extracellular matrix deposition [51]. Currently, there is a dearth of comprehensive investigation into the underlying mechanisms governing the hub genes identified in this study with respect to their involvement in immune cell infiltration during liver fibrosis. Future research endeavors are imperative to unravel their precise modes of action.
In this study, we employed a comprehensive bioinformatics approach to identify key regulatory genes and pathways associated with liver fibrosis. However, the key genes and signaling pathways obtained require further functional validation to strengthen their persuasiveness. Additionally, using rat samples rather than human samples may limit our ability to detect gene expression differences in patients with liver fibrosis.
Conclusion
In summary, this study has identified four key genes, namely Cftr, Cldn4, Map2, and Spp1, which play a crucial role in the progression of liver fibrosis. These genes have the potential to serve as biomarkers for predicting and treating liver fibrosis, as well as providing a solid foundation for further investigation into the pathogenesis and therapeutic strategies of this condition.
Funding
The present study was financially supported by the Yunnan Provincial Department of Science and Technology Plan Project (2018FH001-074) and The Fourth Batch of “Spring City Plan” High-level Talent Program – Young Outstanding Talent Sub-program.
Data availability
The dataset utilized in this study is available in an online database. The specific name of the database and its corresponding accession number can be accessed at:
Supplementary data
The supplementary files are available to download from https://dx-doi-org.web.bisu.edu.cn/10.3233/THC-241142.
Footnotes
Acknowledgments
The authors have no acknowledgments.
Conflict of interest
The authors declare that they have no conflict of interest.