
Abstract
A method for urinary iodine diagnostics using a portable energy-dispersion X-ray fluorescent (EDXRF) spectrometer is proposed. The principle of the method consists in excitation of the sample atoms fluorescence by the energy spectrum intentionally formed from the spectrum of an X-ray tube using an optimized X-ray scheme. The optimization by the criterion of the minimum detection limit for L-series iodine fluorescence lines included the calculation of optimal atomic numbers for materials of a re-radiator and a filter, and their thicknesses, the collimation system parameters, and an X-ray tube voltage. Experimentally achieved detection limits were 45 and 75 μg/L by I-Lα and I-Lβ lines, respectively, at the theoretically extreme value 30 μg/L. This sensitivity is found to be sufficient for urinary iodine diagnostics in the range from 50 to 200 μg/L. The results obtained from different patients have shown the satisfactory convergence for the iodine concentration determination by Lα and I-Lβ fluorescence lines. The simple sample preparation procedure and comparably small sizes of the apparatus allows rapid researches directly in clinical settings.
Introduction
Iodine is an essential element of human nutrition. Nearly a third of the global population has insufficient iodine intake and is at risk of developing Iodine Deficiency Disorders. Most countries have iodine supplementation and monitoring programs. Urinary iodine is the biomarker used for epidemiological studies; only a few methods are currently used routinely for analysis. These methods apply quite expensive instrumentation: high-power X-ray sources [1, 2], inductively coupled plasma mass-spectrometry, instrumental nuclear activation analysis [3], or these are based on the destructive sample enrichment. Such the sample preparation requires time and efforts because possible artifacts may result in non-reproducibility of the measurement results [4]. Therefore, the destructive sample enrichment is carried out in specialized laboratories with qualified personnel. One of inexpensive reliable methods for determination of trace impurities is X-ray fluorescent analysis (XRF).
In XRF classical arrangement, the excitation of sample atoms is realized by the X-ray tube primary spectrum which contains the broad-band bremsstrallung radiation and analytical lines of the target material. Therefore, the X-ray spectrum of the sample shows not only fluorescence lines of its atoms, but also the broad-band background of the primary radiation which is scattered at the sample. This background limits the sensitivity of XRF analysis. For this reason, the direct XRF determination of iodine in urine is impossible because the iodine concentration is in the range from 50 to 200 μg/L (or from 0.05 to 0.20 ppm) [3]. However, using the simple evaporating procedure, the sample can be enriched by a factor 30, i.e. the iodine content increases up to 1 to 6 ppm due to the water removal. After the water evaporation, the sample is a jelly-like residue containing the carbon constituent. So, this residue strongly scatters the primary X-ray radiation. According our calculations and experiments, the background level of the continuous spectrum scattering is quite high and gives no possibility to determine iodine concentration lower than 10 ppm in these objects. Therefore, the sensitivity of the classical XRF arrangement is found to be insufficient for determination of 1 ppm iodine in the jelly-like residue. In the energy dispersion X-ray fluorescent analysis (EDXRF), the selective excitation of sample fluorescence was used in order to lower the detector charging [5]. For the selective excitation, the X-ray primary radiation is transformed using the secondary targets, filters, etc. In the transformed spectrum, a high intensity sector is formed in the certain energy range for effective excitation of analyzed chemical elements, while the intensity of the rest spectrum is being minimized. The transformation of the primary spectrum always decreases the intensity of the analytical lines of sample impurities.
However, lowering the broad-band background sharply increases the contrast (peak-to-background ratio) which defines the sensitivity of the method. Thus, to achieve the maximum sensitivity, it is necessary to calculate optimal parameters of the X-ray optical scheme. The optimization task can be solved for a special range of photon energies where the analytical lines of the impurity are positioned. For this purpose, the calculations are carried out for the distribution of the background caused by the primary radiation scattered by the sample, intensities of the sample fluorescence lines, and artifact peaks [5, 6]. The solution of the optimization inverse problem consists in the selecting and varying the materials and thicknesses of re-radiators and filters, as well as the configuration of the collimation system for obtaining the minimum detection limit [6]. This optimization allows increasing the sensitivity by a factor 50 [7]. It is sufficient for determination of iodine in the jelly-like organic residue which contains about 1–6 ppm of iodine (taking into consideration the thirtyfold enrichment after the water evaporation).
For revealing and separating the fluorescence Ca-Kβ, I-Lα and I-Lβ lines, the computer processing of the X-ray fluorescence spectrum was applied. In the literature, mathematical methods for treatment of the spectra are well known [8]. However, these are unacceptable for our problem, because when the whole spectrum is treated, the contribution of high-intensity lines into the residual functional is dominant. Thus, it is impossible to reveal the intensity variations of weak lines. Therefore, for the processing we selected a narrow spectral area in the photon energy range from 3.8 to 4.4 keV where the strong lines were absent. Such the processing has allowed decreasing the approximation error to 3–5%. The objective of the work was the development of X-ray fluorescent method for rapid diagnostics of iodine in urine.
Sample preparation and investigation technique
The sample preparation procedure
The first stage of the preparation procedure of samples for X-ray fluorescent analysis is their enrichment by iodine by means of water evaporation. Because of the high partial pressure of iodine vapor, fractional volatilization of iodine is possible. So, it is important to choose an optimal temperature of evaporation. The evaporation temperature was chosen by the experimental dependence of I-Lα (E = 3.15 keV) and I-Lβ (E = 4.22 keV) fluorescence lines intensities on the evaporation temperature in comparison with their intensities at 20°C for the same sample. 20% decrease of iodine fluorescence lines intensities was revealed after evaporation at 80°C. In the temperature range from 30 to 40°C no decrease was revealed, so, this range was taken as optimal for the sample iodine enrichment. The samples contain a substantial quantity of calcium, and the strong fluorescence line Ca-Kβ (E = 4.01 keV) is found to be close to the iodine line I-Lα (E = 3.93 keV) in the spectrum. Energy resolution of the detector is insufficient to separate these lines. In order to remove calcium from the sample, we applied the treatment by Sulkovich drug [10]. That has allowed lowering the calcium concentration by about four orders.
Preparation of calibration mixtures
Calibration mixtures were prepared on the base of the same sample of a biological liquid by addition of KI water solutions with different iodine concentrations (“the method of additions” [11]). The first mixture (zero) had the following composition: 0.8 g of urine, 0.1 g of Sulkovich drug, and 0.1 g of distilled water. To increase the iodine concentration, the next three mixtures were prepared by addition 0.1 g of KI water solution with various iodine content (1000, 2000 H 3000 μg/L) to the “zero” composition instead of the distilled water. As a result, we obtained four calibration mixtures with additional iodine concentrations: 0, 100, 200, and 300 μg/L [11]. The calibration mixtures and the experimental samples were placed on Ultraline 10 film and dried at T = 30–40°C in the dustproof chamber to obtain the jelly-like residue. Under such the procedure, the composition of the dried sample filler changes little, and the linear calibration can be used for determination of concentration sensitivity of measurements by the lines I-Lα and I-Lβ independently.
Measurement technique
The X-ray fluorescence spectra of the prepared samples were recorded using a portable EDXRF spectrometer “SPRUT-K” (JSC “Ukrrentgen”, Ukraine) with a SDD detector X-123 (Amptek, USA) and an X-ray tube of 15 W with Ag anode. The time of a spectrum accumulation was 3600 s. The X-ray optical scheme was optimized to provide the maximum sensitivity for revealing the iodine I-Lα and I-Lβ lines in the matrix consisted of Na, K, Cl, P, S, and carbon constituents. The mass portions of the main components of the matrix were determined by XRF, and the carbon portion – by the ratio of Compton and Rayleigh scattering peaks [6]. Optimization parameters were: the material of the secondary target; the material and the thickness of the filter; radiation incident and exit angles; aperture of collimation system; and the thickness of the jelly-like sample. As a result of the optimization procedure, the following set was used: the chromium re-radiator; the incident angle 24±5° for the sample; a 4 μm vanadium filter for the secondary emission and a Soller collimator positioned directly in front of the detector.
Results and discussion
The experimental spectrum of the calibration sample with 300 μg/L iodine (Fig. 1) shows the fluorescence lines of chemical elements of the jelly-like residue and the secondary target lines scattered by the sample (Cr-Kα, Cr-Kβ). The calcium lines are absent in the general spectrum as a result of the sample treatment by Sulkovich drug. In the energy range from 3.8 to 4.3 keV near the lines of interest I-Lα (E = 3.93 keV) and I-Lβ (E = 4.22 keV), a quite low background was achieved only twice exceeding the minimum value calculated theoretically. The intensities of iodine analytical lines correspond to the calculated ones. The obtained contrast level of the iodine lines gives the possibility to determine the iodine concentrations less than100 μg/L in biological liquids using calibration mixtures. The iodine content can be determined independently by the I-Lα and I-Lβ lines. Note, that in the second case the concentration sensitivity of the calibration function will be lower because of twice lower integral intensity of I-Lβ line. However, the stronger I-Lα overlaps the Ca-Kβ line − from the residual calcium after Sulkovich treatment. The positions of I-Lα (E = 3.93 keV) and Ca-Kβ (E = 4.01 keV) lines are differed by energy by 0.075 keV, and their experimental half-width is about 0.130 keV in our spectrum. Thus, these lines can be separated only by the method of the full-profile analysis for the spectral range from 3.8 to 4.1 keV. For such separation, Gaussian profiles were taken for each line. The Gaussian curve half-width was determined by the spectra of standards. The separation was carried out within the multiplet.

A fragment of the spectrum recorded for the calibration sample treated by the Sulkovich drug. Iodine concentration is near 300 μg/L. In the insert: fragments of the experimental spectrum (curve 1), and the calculated (curve 2) in the photon energy range from 3.8 to 4.4 keV.
The adequacy of the separation is confirmed by the results of the mathematical processing the spectra of calibration mixtures different by iodine concentration but similar by the quantity of the residual calcium (Fig. 2). As it is seen from the figure, the intensity of Ca-Kβ line remains practically unchanged as the iodine concentration grows in the calibration mixture, while the intensity of the separated I-Lα line increases proportionally to the iodine concentration.
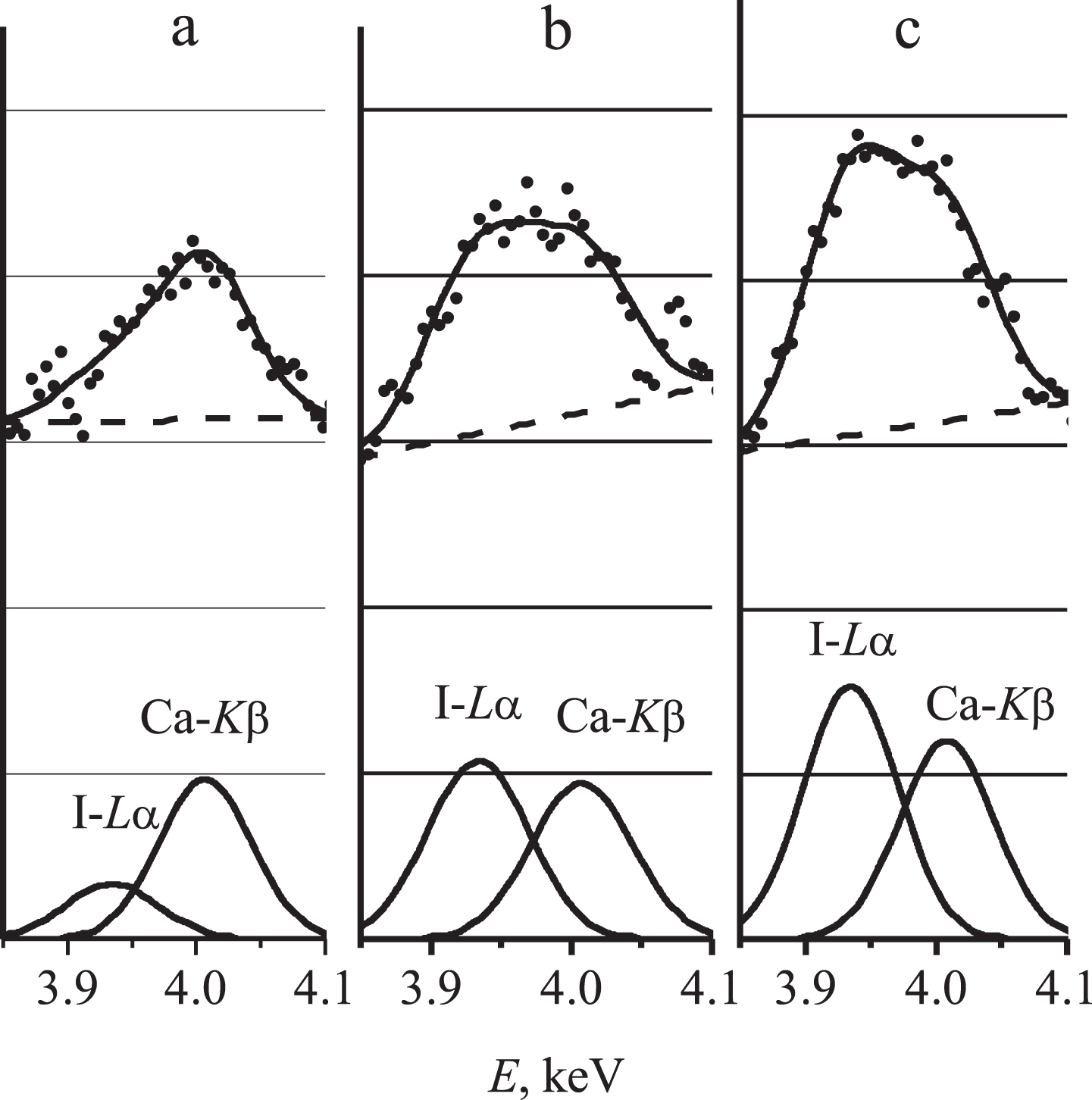
Separation of superimposed I-Lα and Ca-Kβ lines in the spectra of calibration mixtures with different iodine content: a – without additional iodine; b – with 200 μg/L additional iodine; c – with 300 μg/L additional iodine.
For quantitative determinations of iodine concentrations by I-Lα intensities, the calibration mixtures based on the urine samples from different patients were used (Fig. 3). The plot 1 corresponds to the urine samples with “low” iodine contents (from 100 to 110 μg/L), and the plot 2 is based on the samples with “high” iodine concentration (about 180 μg/L). Naturally, these plots are shifted from one another by the Y-axis, but have the same slopes. From the slope, the experimental value ∂ N / - ∂ C= 300 counts/(100 μg/L) corresponding to the theoretically calculated one was determined.
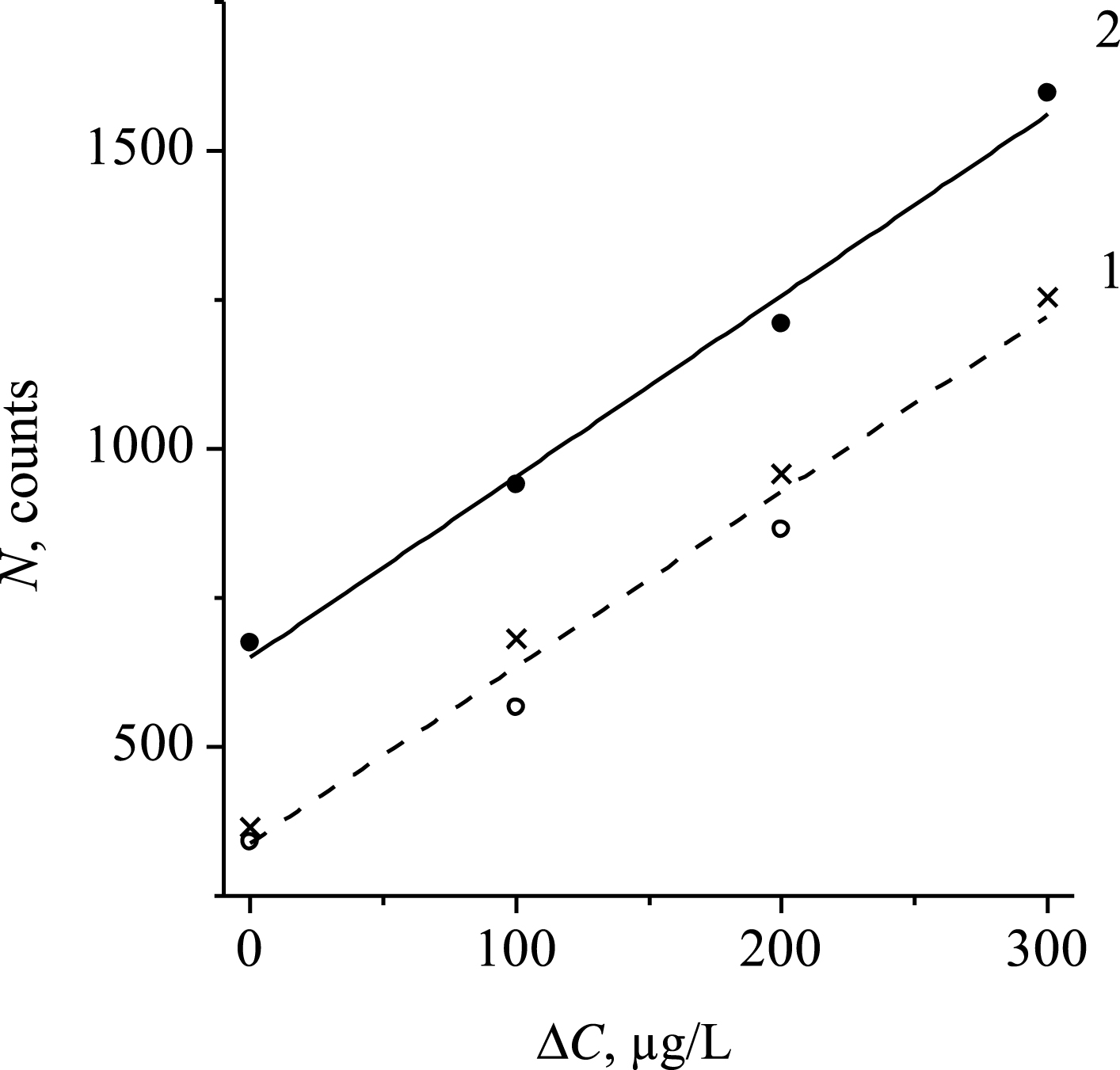
The dependences of I-Lα line integral intensity on the additional iodine mass portion, ΔC, in the calibration mixture: 1 – iodine concentration in the initial sample is 100 μg/L; 2 – iodine concentration in the initial sample is 180 μg/L.
Both for the experimental and the theoretical curves, the detection limit is calculated [11] as (Fig. 1):
The integral intensity of the background, N b , can be determined by the area bounded by the background line under the experimental curve (Fig. 1). Then, , where is the average accumulation of counts per a channel, n is the number of channels over which the summation takes place to measure the integral intensity. Taking into account = 140 counts, and n = 15 channels, we obtain the detection limit Cmin = 45.8 μg/L for the I-Lα line. The theoretical detection limit is Cmin = 32 μg/L at = 70 counts/channel (Fig. 1). Thus, the experimentally achieved sensitivity of analysis is only by a factor 1.5 worse than the highest of the theoretically possible values. For I-Lβ line with = 170 counts/s and ∂ N / - ∂ C= 200 counts/(100 μg/L) we obtain Cmin = 75.7 μg/L. The achieved sensitivity level is sufficient for iodine in urine diagnostics in the iodine concentration range from 50 to 200 μg/L.
The results of clinical investigations for a group of patients are given in Table 1. If for evaluation of the detection limit we used integral intensities (according to the expression for Cmin), here we give peak intensities for visibility of comparison with Fig. 1. It is seen that patients 3, 4, and 5 have the lowered iodine concentration, and patients 2 and12 – evidently heightened level in comparison with a normal level (about 100–150 μg/L).
Iodine fluorescence I-Lα and I-Lβ peak intensities, and the results of determination of iodine concentrations in urine from different patients by these lines independently. Concentration sensitivity by the peak intensity is 40 counts/(100 μg/L) for I-Lα line, and 26 counts/(100 μg/L) for I-Lβ line
The measurements of the urine samples obtained from different patients confirm the not bad convergence of the results of the iodine independent determinations by I-Lα and I-Lβ line intensities. Using such measurements it is possible to obtain quite rapidly the information not only about the iodine concentration in the patient’s urine, but also on its variation during the patient treatment. The increase of the accuracy of the measurements can be achieved due to improving the statistics of counts accumulation, for example, with increasing the X-ray source power and the optimized X-ray optic scheme.
The proposed method of the rapid diagnostics of iodine in urine illustrates the possibilities for increasing the sensitivity of X-ray fluorescent analysis for solution of a special analytical task. The principle of the method consists in fluorescence excitation of sample atoms using a specially formed spectrum separated from the spectrum of an X-ray tube. Optimization of the apparatus X-ray optic scheme, namely, calculations of atomic numbers and thicknesses of a re-radiator and a filter, as well as parameters of a collimation system, - was carried out to provide the minimum detection limit for I-Lα and I-Lβ fluorescence lines in the urine dried sample. Experimentally achieved detection limits are 45 μg/L (for I-Lα) and 75 μg/L (for I-Lβ) at the calculated minimum value 30 μg/L. The high sensitivity of the method and the possibility to determine the iodine concentration by I-Lα and I-Lβ fluorescence lines independently allows carrying out the rapid diagnostics of iodine in urine in the range from 50 to 200 μg/L. Small sizes of the apparatus and the simplicity of the sample preparation procedure open perspectives for application of the method under clinic conditions. Analogous solutions are promising for increasing the sensitivity of X-ray analysis in biology and medicine areas, as well as for materials where a strongly scattering component is difficult to remove.