
Abstract
Background:
2′-C-methyl and 4′-azido nucleosides have previously demonstrated inhibition of hepatitis C virus (HCV) replication by targeting the RNA-dependent RNA polymerase NS5B. In an effort to discover new and more potent anti-HCV agents, we envisioned synthesizing nucleoside analogues by combining the 2′-C-methylmoiety with the 4′-azido-moiety into one molecule.
Methods:
2′-C-methyl-4′-azido pyrimidine nucleosides were synthesized by first converting 2′-C-methyl ribonucleosides to the corresponding 4′-exocyclic methylene nucleosides. Treatment with iodine azide, benzoylation of the 2′- and 3′-hydroxy groups, oxidative displacement of the 5′-iodo group with meta-chloroperoxybenzoic acid, and debenzoylation gave the desired 2′-C-methyl-4′-azido uridine and thymidine analogues in good yield. Standard conversion of uridine to cytidine via the 4-triazole yielded 2′-C-methyl-4′-azido cytidine. In addition, 5′-phosphoramidate derivatives of 2′-C-methyl-4′-azido uridine and cytidine were synthesized to bypass the initial phosphorylation step.
Results:
The prepared nucleosides and their 5′-monophosphate prodrugs were evaluated for their ability to inhibit replication of the hepatitis C virus in a subgenomic replicon cell based assay. Cytotoxicity in Huh7 cells was determined simultaneously with anti-HCV activity by extraction and amplification of both HCV RNA and ribosomal RNA. Among the newly synthesized compounds, only the 5′-monophosphate nucleoside prodrugs had modest and selective anti-HCV activity. All prepared pyrimidine nucleosides and 5′-monophosphate nucleoside prodrugs displayed no evidence of cytotoxicity at high concentrations.
Conclusions:
This work is the first example of both inactive uridine and cytidine analogues of a nucleoside being converted to active anti-HCV nucleosides via 5′-monophosphate prodrugs.
Introduction
Twenty years ago, hepatitis C virus (HCV) was discovered by scientists at Chiron (now Novartis) and the Centers for Disease Control and Prevention to be the causative agent of non-A and non-B hepatitis in humans. Today, this virus has infected an estimated 3% of the global population and 3–4 million individuals become newly infected each year [1,2]. HCV infections often lead to reduced liver function, cirrhosis, and hepatocellular carcinoma, and eventually liver transplantation. The current approved therapy based on pegylated interferon alone or in combination with ribavirin is effective in only approximately half of the genotype 1 population [3,4]. Moreover, this limited efficacy is often associated with significant side effects leading to discontinuation of treatment [5–7]. Therefore, there is a need for the development of more effective therapeutic agents for the treatment of HCV infection.
The HCV RNA-dependent RNA polymerase (NS5B) and NS3/4A protease are currently the most promising targets for the development of novel treatments [8–11]. The activity of these virally encoded enzymes is essential for HCV replication, and antiviral agents targeting these enzymes are in both preclinical and clinicaldevelopment. To date, most of the reported nucleoside analogues that inhibit HCV polymerase have modifications at the 2′ or 4′ positions of the sugar [1–12]. Several 2′-C-methyl nucleoside analogues [13–16] and 4′-azido cytidine analogues [17,18] have been identified as potent inhibitors of HCV NS5B polymerase (Figure 1).
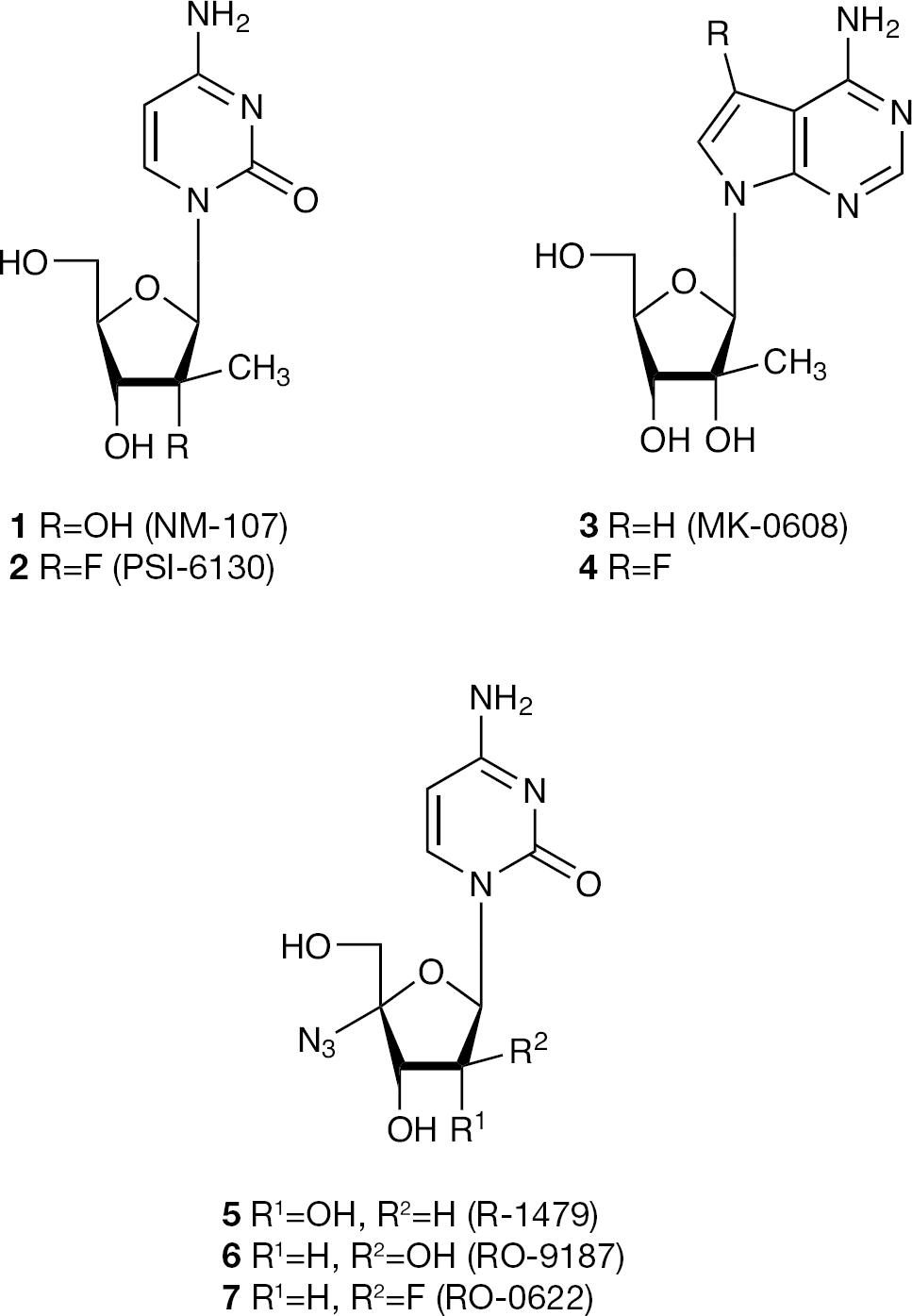
Structures of 2′-C-methyl and 4′-azido nucleosides
In an effort to discover new and more potent anti-HCV agents, we envisioned synthesizing a nucleoside analogue by combining the 2′-C-methyl moiety of NM-107 (
Methods
Chemistry
Thin layer chromatography was carried out on precoated silica gel thin layer sheets 60 F254 from EMD Merck (Darmstadt, Germany). Plate layer chromatography (PLC) from Analtech, Inc. (Newark, DE, USA) was employed for purification of products. 1H NMR (400.14 MHz) was recorded on Varian VNMR 400 spectrometer (Varian, Inc., Palo Alto, CA, USA). Mass spectral analyses were performed on a Micromass TOF instrument (Waters Corp., Milford, MA, USA) and HPLC (Hewlett–Packard, Palo Alto, CA, USA) driven electrospray MS instrument. Analytical HPLC analyses were performed on a Hewlett–Packard HPLC with a Phenomenex Gemini-NX column (2×50 mm, 3 μm, C18, 110 Å). Mobile phase flow was 0.7 ml/min with a 3.5 min gradient from 96% aqueous media (0.05% formic acid) to 96% CH3CN (0.05% formic acid) with a 5.5 min total acquisition time and 190–360 nm photodiode array detection).
1-(2-Methyl-5-iodo-β-D-ribofuranosyl)uracil (9a) and 1-(2-methyl-5-iodo-β-D-ribofuranosyl)thymine (9b)
2′-C-Methyluridine (5 g, 200 mmol), triphenylphosphine (8 g, 307 mmol) and imidazole (2.09 g, 307 mmol) were slurried in anhydrous tetrahydrofuran (THF). A solution of I2 (5.7 g, 220 mmol) in THF was added slowly to the slurry while the reaction temperature was maintained below 28°C. The reaction mixture was stirred at room temperature for 18 h. The reaction was quenched with water and extracted with ethyl acetate. After evaporation of the solvent, the residue was purified by column chromatography using 5% methanol in dichloromethane as eluent to obtain
In a similar manner from
1-(4-Azido-2,3-O-dibenzoyl-2-methyl-5-iodo-β-D-ribofuranosyl) uracil (12a) and 1-(4-azido-2,3-O-dibenzoyl-2-methyl-5-iodo-β-D-ribofuranosyl)thymine (12b)
A solution of

Synthetic route to 2′C-methyl-4′azido pyrimidine nucleosides
1-(4-Azido-5-O-(4-chloro)benzoyl-2,3-O-dibenzoyl-2-methyl-β-D-ribofuranosyl)uracil (13a) and 1-(4-azido-5-O-(4-chloro)benzoyl-2,3-O-dibenzoyl-2-methyl-β-D-ribofuranosyl) thymine (13b)
To a solution of compound
1-(4-Azido-2-methyl-β-D-ribofuranosyl)uracil (14a) and 1-(4-azido-2-methyl-β-D-ribofuranosyl)thymine (14b)
Compound
1-(4-Azido-2-methyl-β-D-ribofuranosyl)cytosine (15)
To a solution of
4-Azido-2-C-methyluridine-5′-O-[Phenyl(ethyloxy-l-alaninyl)]phosphate (16) and 4-azido-2-C-methylcytidine-5′-O-[phenyl(ethyloxy-l-alaninyl)] phosphate (17)
To a solution of
Virology
HCV replicon assays
Huh 7 clone B cells containing HCV replicon RNA were seeded in a 96-well plate at 5,000 cells/well, and the compounds tested initially at 10 μM in triplicate immediately after seeding. Following 5 days incubation (37°C, 5% CO2), total cellular RNA was isolated by using the VersaGene RNA purification kit (Gentra, Minneapolis, MN, USA). Replicon RNA and an internal control (TaqMan rRNA control reagents, Applied Biosystems, Foster City, CA, USA) were amplified in a single step multiplex real time reverse transcriptase (RT)-PCR assay. A dose–response curve was determined for nucleosides demonstrating antiviral activity below 10 μM. The antiviral effectiveness of the nucleosides was calculated by subtracting the threshold RT-PCR cycle of the test compound from the threshold RT-PCR cycle of the no-drug control (ΔCt HCV). A ΔCt of 3.3 equals a 1 log10 reduction (equal to 90% less starting material) in replicon RNA levels. The cytotoxicity of the compounds was also calculated by using the ΔCt ribosomal RNA (rRNA) values. (2′-Me-C) was used as the control. To determine 90% effective concentration, 50% effective concetration and 50% cytotoxicity concentrations, ΔCt values were first converted into fraction of starting material and were then used to calculate the percentage inhibition.
Results
During the preparation of this paper, an alternate synthesis appeared for 2′-C-methyl-4′-azido cytidine (
2′-C-Methyl ribonucleosides
The 2′-C-methyl-4′-azido nucleosides
Anti-HCV replicon activity and cellular toxicity of synthesized nucleoside and nucleotide analogues a
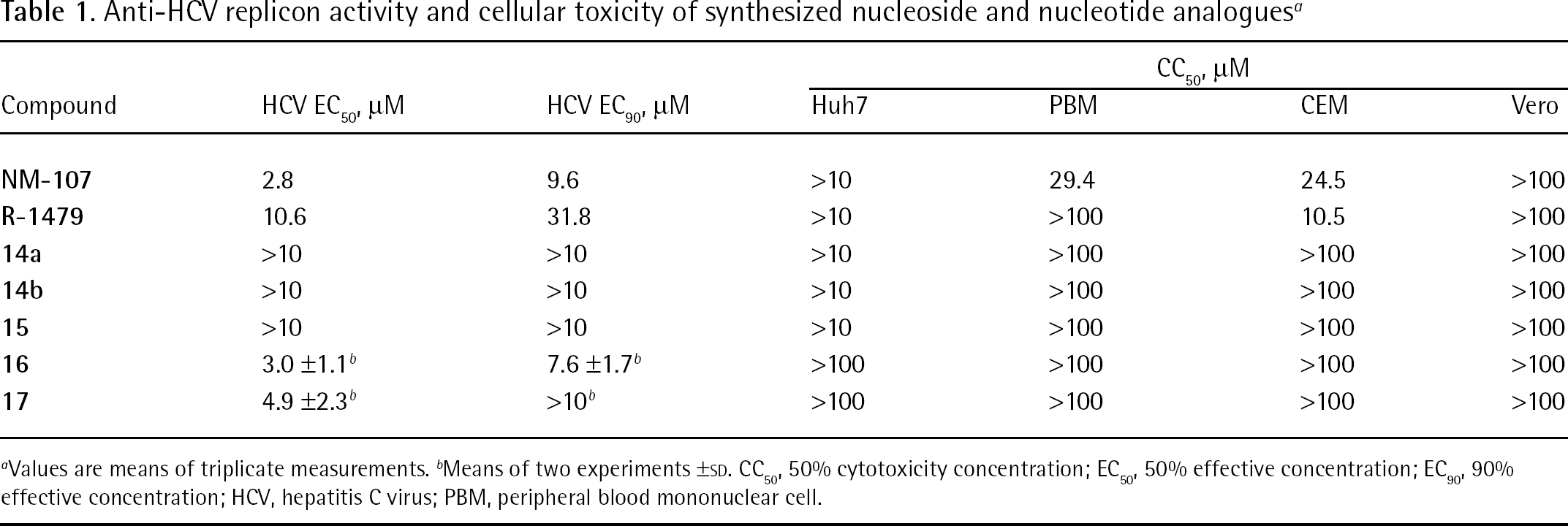
Values are means of triplicate measurements.
Means of two experiments ±SD. CC50, 50% cytotoxicity concentration; EC50, 50% effective concentration; EC90, 90% effective concentration; HCV, hepatitis C virus; PBM, peripheral blood mononuclear cell.
Because the cytidine analogue
The target monophosphate prodrugs
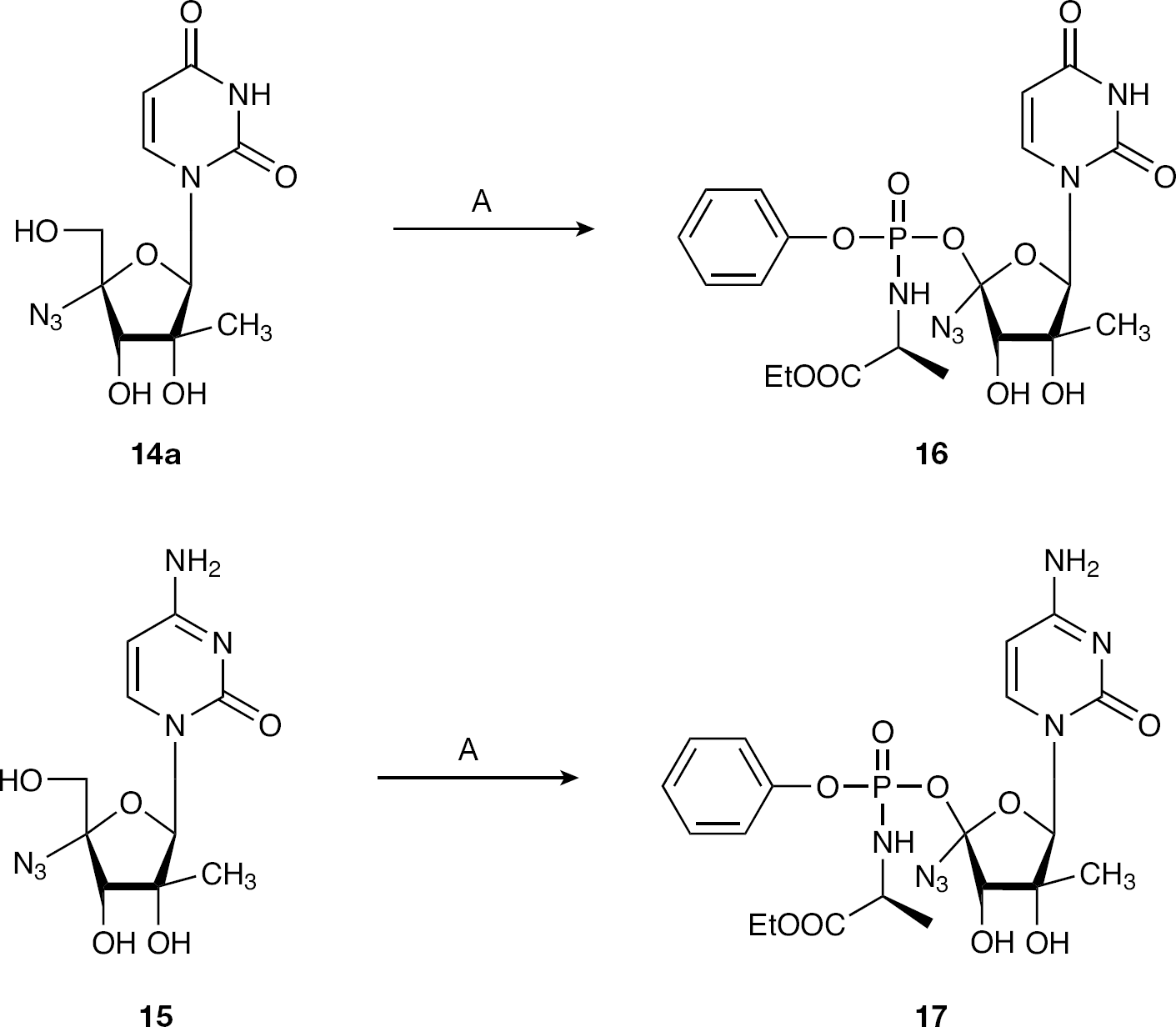
Reagents and conditions
In contrast to the parent nucleosides
Discussion
Three 2′-C-methyl-4′-azido-pyrimidine nucleosides and two novel 5′-monophosphate prodrugs were synthesized and evaluated for their antiviral activity in an HCV replicon system. As expected, 2′-C-methyl-4′-azido cytidine
Footnotes
Acknowledgements
This work is supported in part by NIH grants 2P30-AI-50409 (CFAR), 5R37-AI-041980, and by the Department of Veterans Affairs.
RFS is the principal founder and a Director of RFS Pharma, LLC. His laboratory received no funding from RFS Pharma for this work. All other authors declare no competing interests.