
Abstract
Background:
Amantadine constitutes an interesting, diamond crystal lattice-shaped, antivirally active amine with an inhibitory effect on influenza A viruses causing common ‘flu’ in humans. Unfortunately, amantadine forfeited most of its therapeutic potential because of resistance development in recent influenza A virus isolates. The antiviral efficacy of amantadine congeners can be chemically modified, resulting in re-constitution, improvement and/or extension of antiviral activities mediated by amino-adamantyls.
Methods:
Newly synthesized compounds were evaluated towards HIV type-1 (HIV-1) replication in primary human lymphocytes. One N-phenacyl amantadine derivative was investigated for inhibiting the in vitro replication of respiratory viruses (influenza A viruses, influenza B virus, human parainfluenza virus type 3 and severe acute respiratory syndrome coronavirus).
Results:
Two ketone-stabilized 1-adamantyl singlet nitrenes were discovered serendipitously. To our best knowledge these are the first persistently stable nitrenes to be reported. Their structure was proved by determining the X-ray single crystal structure of one hydrolytic elaboration product. This salt adduct revealed an incommensurately modulated crystal structure, which was solved by extensive computational refinement. We could show that ketone-stabilized 1-adamantyl singlet nitrenes are versatile synthons for the synthesis of antiviral drug candidates. An amantadine–folate conjugate was inhibitory on HIV-1 replication in primary human lymphocytes, and one N-phenacyl amantadine derivative was inhibitory towards low pathogenic avian influenza A virus (H5N1) replication in vitro.
Conclusions:
These results indicate that the aromatic-aliphatic ketone-stabilized 1-adamantyl singlet nitrenes, beyond being of fundamental interest in organic chemistry, represent versatile synthons for the synthesis of new amantadine-related potentially antiviral drugs.
Introduction
Amantadine (tricyclo[3.3.1.13,7]decan-1-amine, adamantan-1-amine; 1), a tricyclic amine with diamondoid structure [1], was discovered to be efficacious as a pharmacological treatment of infections with certain Orthomyxoviridae, notably influenza A viruses, in 1964 [2], soon after its first synthesis reported by Stetter et al. [3] in 1960. However, today 1 is considered obsolete as a treatment of human influenza A virus infections (common ‘flu’), since most of recently (2006–2011) circulating influenza A virus isolates were found to be resistant to 1 and its more potent congener rimantadine ([±]-α-[1-adamantyl]ethanamine) [4,5]. The mechanism-of-action by which 1 and rimantadine (as well as other cyclic amines like cyclooctanamine or bornan-2-amines) [6] suppress influenza A virus replication is blockage of the influenza A virus M2 protein transmembrane proton channel [7–9]. The latter is encoded by the seventh segment of the eightfold segmented positive single-stranded RNA genome of the enveloped influenza A virus [9]. The M2 block by 1 and its congeners impairs pH-dependent uncoating of influenza A viruses by low pH-triggered haemagglutinin (HA)-initiated virus envelope-endosomal membrane fusion [9]. The antiviral efficacy of amantadine congeners can be chemically modified, resulting in re-constitution, improvement and/or extension of antiviral activities mediated by amino-adamantyls [10,11]. Therefore we felt encouraged to initiate studies on unprecedented chemical transactions of 1.
Materials and methods
Materials
Common laboratory chemicals were of analytical or best available quality and were purchased from AppliChem GmbH (Darmstadt, Germany) and Merck-Schuchardt OHG (Hohenbrunn, Germany). 2-Bromo-4′-fluoroacetophenone (purity according to manufacturer ≥96.5% [gas chromatography (GC) area %]; actual purity 98.2% [GC area %]) and 2-bromo-4′-methoxyacetophenone (purity according to manufacturer ≥96.5% [GC area %]; actual purity 99.6% [GC area %]) were purchased from Sigma–Aldrich Corporation (St Louis, MO, USA). 1-Adamantanammonium chloride (international non-proprietary name: amantadine hydrochloride; purity >99% [argentometric titration]) was purchased from Merck–Schuchardt OHG. Adenine (purity >99% [HPLC]), L-folic acid dihydrate (purity according to manufacturer ≥96% [HPLC]; actual purity 98.5% [n/n] on dry basis as determined by 1H NMR), cytidine (purity ≥99% [HPLC]) and anhydrous N,N-dimethylformamide (DMF; residual water ≤0.01% [Karl Fischer titration]) were purchased from AppliChem GmbH.
Methods
The Fourier transform infrared spectroscopy (FT–IR) experiments were recorded in solid potassium bromide pellets on a Digilab Excalibur FTS 4000 spectrophotometer (Digilab Inc., Holliston, MA, USA) at Currenta GmbH & Co. OHG (Leverkusen, Germany). Given FT–IR absorbance bands, expressed in wavenumbers (cm−1), are characterized in intensity as strong (str), middle (m), weak (w) and broad (br). The 1H NMR (700.430 MHz), 13C NMR and 13C DEPTQ NMR (176.120 MHz) spectroscopy experiments were recorded at a temperature of 300.0 K using a Bruker Avance 700 NMR spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany) at Currenta GmbH & Co. OHG. The spectra were referenced to the centre of the NMR solvent signal (1H NMR: δ [ppm] 7.27 [CDCl3], 2.52 [(D6) dimethyl sulfoxide (DMSO)], 4.82 [D2O], 7.20, 7.57, 8.72 [(D5) pyridine]; 13C NMR: δ [ppm] 77.02 [CDCl3], 39.52 [(D6) DMSO], 123.98, 135.98, 150.36 [(D5)pyridine]). Given chemical shifts δ (from tetramethylsilane [TMS]: δ=0) are specified as singlet (s), broad singlet (br s), doublet (d), doublet of doublet (dd), doublet of doublet of doublet (ddd), triplet (t), doublet of triplet (dt), quartet (q), doublet of quartet (dq) and multiplet (m). Optical rotations were measured at the fixed temperature of 20 ±0.5°C on a PerkinElmer 241 polarimeter (PerkinElmer Inc., Waltham, MA, USA) equipped with a 100 mm lightpath cell at Currenta GmbH & Co. OHG. Elemental analyses (C, H, N and O) were conducted on the EURO EA3000 CHNS–O elemental analyser (EuroVector SpA, Milan, Italy) by HEKAtech GmbH (Wegberg, Germany).
Chemistry
1-(4-Fluorophenyl)ethanone – tricyclo[3.3.1.13,7]dec-1-ylazanylidene (1:1)x0.11 H2O (3)
2-Bromo-4′-fluoroacetophenone (2) (7.40 g, 34.10 mmol, 1 equivalent), 1-adamantanammonium chloride (international non-proprietary name: amantadine (1) hydrochloride; 6.40 g, 34.10 mmol, 1 equivalent) and sodium hydrogen carbonate (NaHCO3; 5.76 g, 68.56 mmol, 2.01 equivalents) were suspended in anhydrous DMF (200 ml). The mixture was refluxed for 2 h. Then water (200 ml) was added in small portions through the reflux condensor. The yellow-orange solution was heated for an additional 5 min. A yellow crystalline precipitate evolved. The crystallizing suspension was frozen at −25°C for 3 h. The evolved crude product was filtered and dried over CaCl2 in vacuo. From the filtrate (pH 10–11) by freezing at −25°C for 9 h and treatment as before additional substance could be recovered. The finely grounded light yellow crude product (7.03 g) was suspended in acetone (100 ml). The suspension was stirred at room temperature for 20 min. The suspension was additionally frozen at −25°C for 2 h. The evolved precipitate was filtered, washed on the filter with water (2×125 ml), and dried over CaCl2 in vacuo to give 3 (4.59 g, 47%) as yellowish-white powder.
1-(4-Fluorophenyl)ethanone – tricyclo[3.3.1.13,7]dec-1-ylazanylidene (1:1) (4)
Compound 3 (4.59 g, 15.86 mmol, 1 equivalent) and adenine (9H-purin-6-amine; 2.10 g, 15.54 mmol, 0.98 equivalents) were suspended in 50% (v/v) aqueous acetone (520 ml). The suspension was refluxed for 15 min. Afterwards, the mixture was cooled at 0–2°C for 1 h. Then the suspension was frozen at −25°C for 1 h. After adding water (200 ml), the suspension was frozen at −25°C for 2.5 h. The evolved precipitate was filtered, washed on the filter with water (4×125 ml), and dried over CaCl2 in vacuo to give 4 (3.68 g, 81%) as white powder.
[1,4-Bis(4-methoxyphenyl)butane-1,4-dione (7) – tricyclo[3.3.1.13,7]dec-1-ylazanylidene (0.5:1)]x0.25 H2Ox0.25 NaHCO3 (6)
2-Bromo-4′-methoxyacetophenone (5) (7.81 g, 34.10 mmol, 1 equivalent), 1-adamantanammonium chloride (6.40 g, 34.10 mmol, 1 equivalent) and NaHCO3 (5.76 g, 68.56 mmol, 2.01 equivalents) were suspended in anhydrous DMF (200 ml). The mixture was refluxed for 2.5 h. Then, water (100 ml) was added in small portions through the reflux condensor. The yellow-orange solution was heated for an additional 8 min. Afterwards, the suspension was pre-cooled at 0–2°C for 3 h. The crystallizing suspension was frozen at −25°C for 1 h. Then water (100 ml) was added, the suspension (pH 10–11) was shaken and additionally frozen at −25°C for 3 h. The evolved crystalline crude product was filtered and dried over CaCl2 in vacuo. The yellow crude product (9.32 g) was suspended in acetone (30 ml). The suspension was stirred at room temperature for 20 min. The suspension was additionally frozen at −25°C for 2.5 h. The evolved precipitate was filtered, separately washed on the filter with water (2×125 ml), and dried over CaCl2 in vacuo. From the filtrate, additional product could be recovered. The yields were combined to give 6 (3.36 g, 30%) as yellowish-white powder.
[1,4-Bis(4-hydroxyphenyl)butane-1,4-dione-1,4-bis(4-methoxyphenyl)butane-1,4-dione (7) – (2S,2′S)-2,2′-{[(tricyclo[3.3.1.13,7]dec-1-ylamino)methanediyl] bis(pteroylimino)}dipentanedioic acid (0.2:1:1)]x3 H2Ox0.4 acetone (9)
Compound 6 (2.30 g, 7.10 mmol, 1 equivalent) and L-folic acid dihydrate (8) (1.84 g, 3.85 mmol, 0.54 equivalents) were suspended in acetone (115 ml). After adding water (45 ml), the suspension was stirred at room temperature for 8 min. Then acetone (70 ml) was added and the suspension was heated under stirring to 50–60°C for 8 min. Water (45 ml) was added and the mixture was shaken vigorously for 1 min. After adding acetone (70 ml) and water (115 ml), the suspension was refluxed for 35 min. After short pre-cooling, the suspension was filtrated through two layers of filter paper. Residues were transferred with acetone (90 ml). In the filter folic acid remained. The filtrate (approximately 550 ml) was pre-cooled at 0–2°C for 15 min. Then it was frozen at −25°C for 3 h. Afterwards, the volume was reduced in vacuo from approximately 550 ml to approximately 275 ml. The evolved hydrogel was vacuum-filtered (extremely long filtering time of 22 h) and dried over CaCl2 in vacuo to give 9 (1.27 g, 45%) as yellow-orange powder.
Pentasodium (2S,2′S)-2,2′-{azanidediylbis[(4-{[(4-oxo-3,4-dihydropteridine-2,6-diyl)methyl]amino}benzoyl) amino]}dipentanedioatex3 H2Ox2.5 acetone (10)
A solution of NaHCO3 (200 mg, 2.38 mmol, 4.38 equivalents) in water (20 ml) and acetone (10 ml) was prepared. It was stirred until all NaHCO3 had dissolved (10 min). To this solution 9 (800 mg, 542.85 μmol, 1 equivalent) was added by transferring with acetone (10 ml). The suspension was stirred at room temperature for 5 min. Then water (10 ml) was added and the suspension was stirred at 22–24°C for 1 h. During that time the yellow-orange 9 dissolved slowly. Then acetone (10 ml) and water (10 ml) were added and the suspension was frozen at −25°C for 40 min. After adding acetone (20 ml), the suspension was frozen at −25°C for 40 min. The evolved precipitate was filtered, washed on the filter with acetone (2×125 ml), and dried over CaCl2 in vacuo to give 10 (530 mg, 95%) as yellow-orange powder.
Disodium (2S)-2-(pteroylamino)pentanedioatex4 H2O (disodium L-folate tetrahydrate) (11)
A solution of NaHCO3 (300 mg, 3.57 mmol, 2.13 equivalents) in water (20 ml) and acetone (10 ml) was prepared. It was stirred until all NaHCO3 had dissolved (10 min). To this solution L-folic acid dihydrate (8) (800 mg, 1.68 mmol, 1 equivalent) was added by transferring with water (20 ml). The suspension was stirred at 22–24°C for 30 min. During that time the yellow 8 dissolved. Then acetone (20 ml) was added and the suspension was frozen at −25°C for 2 h. The evolved precipitate was filtered, washed on the filter with acetone (125 ml), and dried over CaCl2 in vacuo. The filtrate was frozen at −25°C for 1 h. The additional yield was filtered, washed on the filter with acetone (70 ml), and dried over CaCl2 in vacuo. The yields were combined to give 11 (860 mg, 92%) as yellow-orange powder.
1-(4-Fluorophenyl)-2-(tricyclo[3.3.1.13,7]dec-1-ylamino) ethanone hydrochloridex0.11 H2O (12)
Compound 4 (3.68 g, 12.81 mmol, 1 equivalent) and cytidine (3.18 g, 13.08 mmol, 1.02 equivalents) were suspended in acetone (200 ml) and water (80 ml). Then 10.27 M (32% [m/m]) aqueous hydrochloric acid (1.7 ml, 17.46 mmol, 1.36 equivalents) was added. The suspension was stirred at room temperature for 10 min. Afterwards, it was heated to 50–60°C under stirring for 5 min. After adding acetone (200 ml) and water (40 ml), the mixture was refluxed for 80 min. A lemon yellow suspension resulted, which was hot filtrated through two layers of filter paper. Residues were transferred with acetone (20 ml). The filtrate was frozen at −25°C for 3 h. Afterwards, the volume of the solution (pH 5–6) was reduced from 540 ml to approximately 150 ml by evaporation in vacuo. The suspension was frozen at −25°C for 2 h. The evolved first crop (1.48 g) of the crude product was filtered and dried over CaCl2 in vacuo. The filtrate was frozen at −25°C for 30 min. The evolved second crop (200 mg) of the crude product was filtered and dried over CaCl2 in vacuo. The combined first crude product (1.68 g, 41%) was suspended in acetone (20 ml) and water (10 ml). The suspension was frozen at −25°C for 3 h. The evolved first crop (1.42 g) was filtered and dried over CaCl2 in vacuo. The filtrate was frozen at −25°C for 2 h. The evolved second crop (140 mg) was filtered and dried over CaCl2 in vacuo. The combined second crude product (1.57 g, 38%) was suspended in acetone (10 ml) and water (5 ml), and 10.27 M (32% [m/m]) aqueous hydrochloric acid (0.2 ml, 2.05 mmol) was added. The suspension was frozen at −25°C for 1 h. The evolved first crop of the product was filtered and dried over CaCl2 in vacuo. The filtrate was frozen at −25°C for 1 h. The evolved second crop of the product was filtered and dried over CaCl2 in vacuo. The yields were combined to give 12 (1.57 g, 38%) as yellowish-white crystalline powder.
X-ray crystallography
The X-ray crystallographic determinations of the crystal and molecular structures of the oxidative elaboration product of 3 (3ox) and 7 were performed at a temperature of 97 K (−176.15°C) (3ox) or 110 K (−163.15°C) (7). Crystals of 3ox suitable for analysis were grown by evaporation at room temperature (18–20°C) of the filtrate of a suspension of 3 in 95% (v/v) aqueous ethanol. Crystals of 7 suitable for analysis were grown by cooling to room temperature (18–20°C) of a solution of 6 in 95% (v/v) aqueous ethanol, which was prepared by short boiling. The crystal structure determinations of 3ox and 7 were performed using an Xcalibur series diffractometer (Oxford Diffraction Ltd, Abingdon, UK) equipped with a CCD area detector (model Ruby), a sealed tube with Cu Kα radiation (λ=1.54178 Å), Osmic mirror (Osmic Inc., Auburn Hills, MI, USA) as monochromator and a CryoJet low temperature device. A full sphere of data was collected by φ and ω scans. For data collection and reduction the programme CrysAlis (Oxford Diffraction Ltd, Abingdon, UK) [12] was used. Crystal structure solution was achieved using direct methods as implemented in SHELXTL [13] and visualized using the Bruker XP program [14]. Missing atoms were subsequently located from difference Fourier synthesis and added to the atom list. Least-squares refinement on F2 using all measured intensities was carried out in SHELXTL [13]. All non-hydrogen atoms were refined including anisotropic displacement parameters.
Virology
Biological testing
The biologically employed test compounds were initially dissolved in DMSO at the stock concentration 10 mg/ml, and then appropriately diluted with cell culture medium to achieve a final DMSO content of <1% (v/v) in the cell culture fluid. This DMSO concentration was non-toxic to cells.
HIV-1 replication reverse transcriptase assay
HIV-1LAI [15] was assayed in primary human peripheral blood mononuclear cells (PBMC) in the presence of a drug being evaluated. The parameter for antiviral activity was reduction of reverse transcriptase (RT) activity in the cell supernatant after Triton X–100-mediated lysis of released virions, as measured by 5α-tritiated thymidine 5′-triphosphate ([3H]-dTTP) incorporation into poly(rA)·poly(dT) directed by the primed RNA template poly(rA)·oligo(dT). It should be noted that the assay did not detect RT inhibition by potential RT inhibitors per se, but indirectly quantified the amount of released HIV-1 in the supernatant. The detailed assay methodology was reported previously by Schinazi et al. [16], and was based on an older assay system by Spira et al. [17]. The experiments were conducted in triplicate and treated statistically by regression curve analysis (r2 coefficient of determination equivalent to goodness-of-fit). The RT inhibitor zidovudine (3′-azido-3′-deoxythymidine; Retrovir™, GlaxoSmithKline) served as a positive control. Cytotoxicity on PBMCs exerted by the test compounds was determined as described by Stuyver et al. [18], by application of the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA). Briefly, the phenazine ethosulfate (PES)-coupled reduction of the tetrazolium salt 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) to a purple, water-soluble formazan by living, undamaged cells was measured.
Influenza virus assays of 12
The effect of 12 on influenza infection in vitro was evaluated against A/California/07/2009 (pandemic swine-origin H1N1, which is resistant to amantadine due to an S31N mutation in M2 protein) [19], A/Perth/16/2009 (seasonal H3N2, amantadine-resistance due to S31N mutation in M2 protein) [20,21], A/duck/Minnesota/1525/81 (low pathogenic avian influenza H5N1, amantadine-susceptible, 50% effective inhibitory concentration [EC50; 1 xHCl] 0.54 μg/ml [2.88 μM]) [22,23], A/Vietnam/1203/2004 (highly pathogenic avian influenza H5N1, amantadine-resistance due to L26I and S31N mutations in M2 protein) [24] and B/Florida/4/2006 (BM2 ion channel protein does not contain the amino acid motif targeted by amantadine). Virus was cultured in Madin–Darby canine kidney (MDCK) cells that were maintained in minimal essential medium (MEM) supplemented with 5% fetal bovine serum (FBS). For the in vitro assays, 96-well plates were seeded with 105 MDCK cells/well in MEM (MEM/EBSS; Hyclone, Logan, UT, USA) without FBS or trypsin and supplemented with 50 μg/ml gentamicin. Ribavirin (1-β-D-ribofuranosyl-1H-1,2,4-triazole-3-carboxamide; Virazole™, Copegus™, Rebetol™) served as the positive control for influenza virus assays. The inoculated cell culture infective dose 50% (CCID50) per well was 100 CCID50 (California virus), 20 CCID50 (Perth virus), 25 CCID50 (duck virus), ≥42 CCID50 (Vietnam virus) and 31 CCID50 (Florida virus), yielding a multiplicity of infection (MOI) of ≥0.001 for each influenza virus strain.
Human parainfluenza virus type 3 assay of 12
The effect of 12 on cytopathic effect (CPE) caused by human parainfluenza virus type 3 (isolate 14702; Mononegavirales, Paramyxoviridae, Paramyxovirinae, Respirovirus) was evaluated in MA-104 embryonic African green monkey kidney epithelial cells that were maintained in MEM with 10% FBS. For the in vitro assays 96-well plates were seeded with 4×104 cells/well in MEM with 2% FBS and 50 μg/ml gentamicin. Ribavirin served as the positive control for parainfluenza virus assays. The virus inoculum was approximately 560 CCID50/well yielding an MOI of approximately 0.014 for human parainfluenza virus type 3.
Severe acute respiratory syndrome coronavirus assay of 12
The effect of 12 on severe acute respiratory syndrome (SARS) coronavirus (strain Urbani; Nidovirales, Coronaviridae, Coronavirinae, Betacoronavirus) was evaluated in Vero 76 African green monkey kidney epithelial cells grown in MEM supplemented with 10% FBS. For the in vitro assays, 96-well plates were seeded with 2×104 cells/well in MEM with 2% FBS and 50 μg/ml gentamicin. The positive control was protease inhibitor (4RS)-4-[N-(benzyloxycarbonyl)-L-leucylamino]-6-fluoro-5-oxohexanoic acid N,N-dimethylamide (EP128533; Maxim Pharmaceuticals, San Diego, CA, USA) [25]. The inoculum was approximately 130 CCID50/well, yielding an MOI of approximately 0.007 for SARS coronavirus.
Cytopathic effect and neutral red assays of antiviral effect
The neutral red lysosomal uptake cell viability assay [26] was performed essentially as previously described [27–30]. The 96-well plates were seeded with cells and incubated overnight at 37°C with 5% CO2 before testing. Compound 12 was serially diluted in the prescribed test medium using eight half-log dilutions. Each dilution was added to 5 wells of a 96-well plate with 80–100% confluent cells, and three wells of each dilution were then infected with the test virus. Two wells remained uninfected as toxicity controls. Six wells per plate were set aside as uninfected, untreated cell controls, and six wells per plate were infected with no treatment as virus controls. A known active compound was assayed in parallel as a control. Assay plates were incubated at 37°C with 5% CO2. After the CPE of a virus was observed microscopically in each well and after each well was scored for CPE on a scale of 1–4 with a score of 4 meaning that all cells showed CPE (CPE reduction assay), each well was filled with 0.011% (m/v) neutral red, a vital stain, and the plate was incubated for approximately 2 h at 37°C in the dark. The unincorporated neutral red solution was removed from the wells and the incorporated dye was then eluted by adding Sørensen's citrate-buffered ethanol. The plates were then read on a spectrophotometer at 540 nm to quantify the neutral red taken up by the healthy cells. The optical density of test wells was converted to percentage of cell control and normalized to the virus controls.
The concentration of test compound required to inhibit CPE by 50% or reduce neutral red uptake into cells by 50% (EC50) was calculated by regression analysis. The toxicity of the compound without virus present was similarly calculated using the uninfected wells treated with test compounds compared with untreated cell controls. The concentration of compound that would cause 50% cytotoxic effects in the absence of virus (CC50) was estimated by linear regression analysis. The selectivity index (SI) is the CC50 divided by EC50. Assays were repeated using four 10-fold dilutions to verify inactive results. When antiviral activity was observed, as with the duck influenza strain, the neutral red assay was performed in triplicate with eight half-log dilutions, followed by the virus yield reduction assay in triplicate to confirm and quantify the antiviral activity of 12.
Virus yield reduction assay
The virus yield reduction assay determines actual virus yield in the presence and absence of the test compound; this is the confirmatory assay for antiviral activity. After 3–5 days' incubation when maximum CPE was observed in the assay plates, an aliquot of supernatant fluid was removed from each test well. Replicate wells of each compound concentration or control were pooled and frozen at −80°C. Samples were thawed and diluted by 10-fold serial dilutions. A 100 μl aliquot of each dilution was then plated onto 4 replicate wells of 96-well plates seeded with the applicable cells for each virus strain. Plates were incubated as noted above until viral CPE reached its end point, then each well was scored microscopically for the presence of viral CPE. The virus titre was determined based on the end point using the Reed–Muench method [31]. Test wells were compared with virus control wells, and the concentration of compound required to reduce virus yield by 90% or 1 log10 (EC90) was calculated by regression analysis.
Results
The synthesis of the stabilized singlet nitrenes 3, 4 and 6
During our studies aimed at modifying the antiviral activity of 1 (Figure 1) by chemical transactions, the unprecedented formation of 1-adamantyl nitrene was discovered (Figure 1). In detail, 1 xHCl was refluxed with 2-bromo-4′-fluoroacetophenone (2) in anhydrous DMF under the presence of NaHCO3. The formation of the N-(4-fluorophenacyl)-1 was expected. However, no N-(4-fluorophenacyl)-1 could be isolated, and instead a yellowish-white powder 3 was obtained (Figure 1). An analogous reaction of 1 xHCl with 2-bromo-4′-methoxyacetophenone (5) produced the yellowish powder 6 (Figure 1). The compound 3 was purified into a white powder 4 by refluxing with the weak nucleobase adenine (pKa 4.22 at 293 K) [32], and 4 was analysed by proton nuclear magnetic resonance spectroscopy (1H NMR; Figure 2A) and by carbon-13 (13C) distortionless enhancement by polarization transfer including detection of quaternary nuclei (DEPTQ) [33] nuclear magnetic resonance spectroscopy (NMR) in deuterated pyridine ([D5]pyridine). A reproduced batch of 4 was accordingly investigated in [D5]pyridine by 1H NMR and 13C NMR. Finally, 4 was also examined in deuterated chloroform (CDCl3) by 1H NMR (Figure 2B) and 13C DEPTQ NMR, and by Fourier transform infrared spectroscopy (FT–IR). For all listed, but not illustrated additional spectra see Additional file 1.
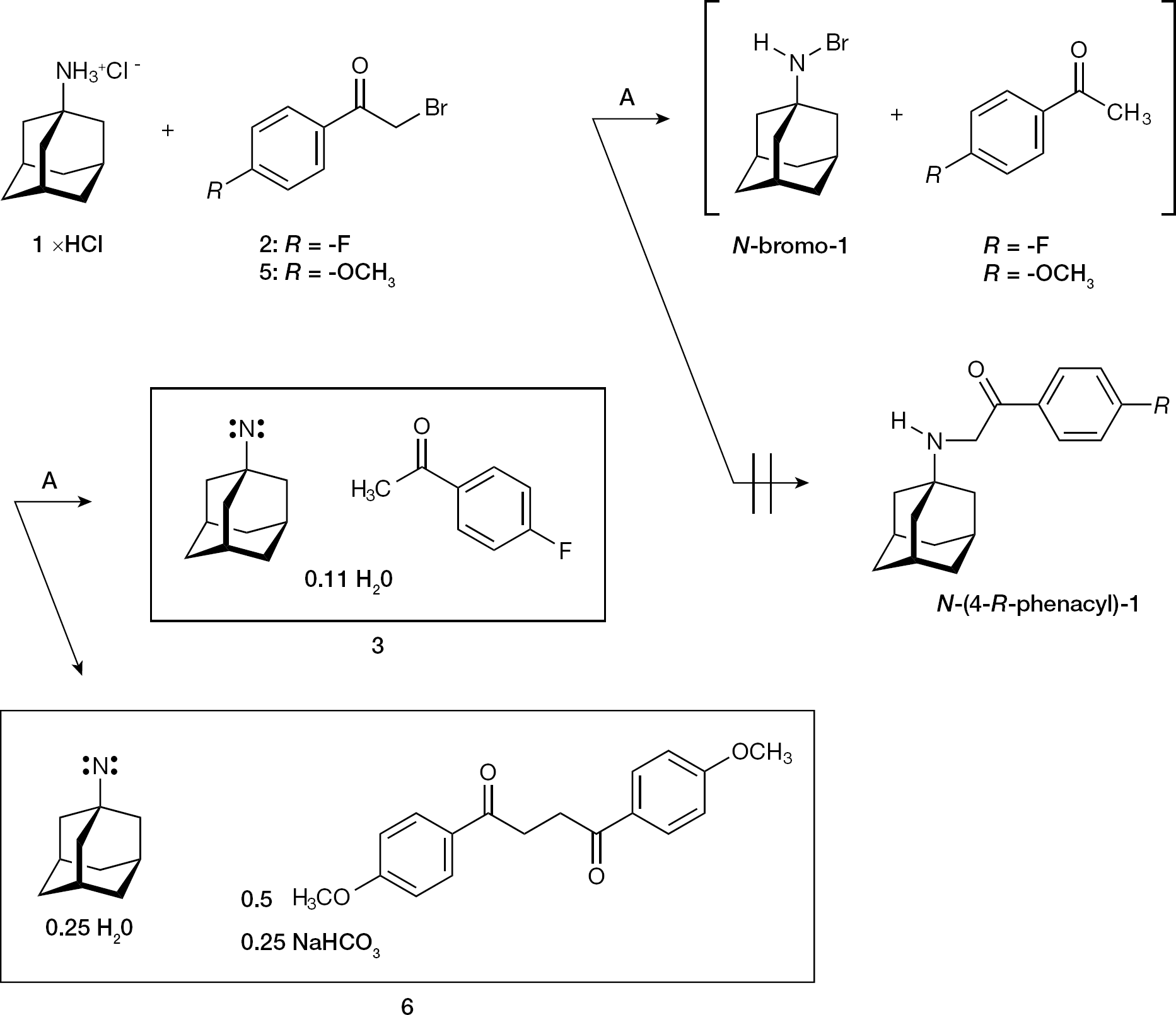
The formation of 1-adamantyl singlet nitrene adducts
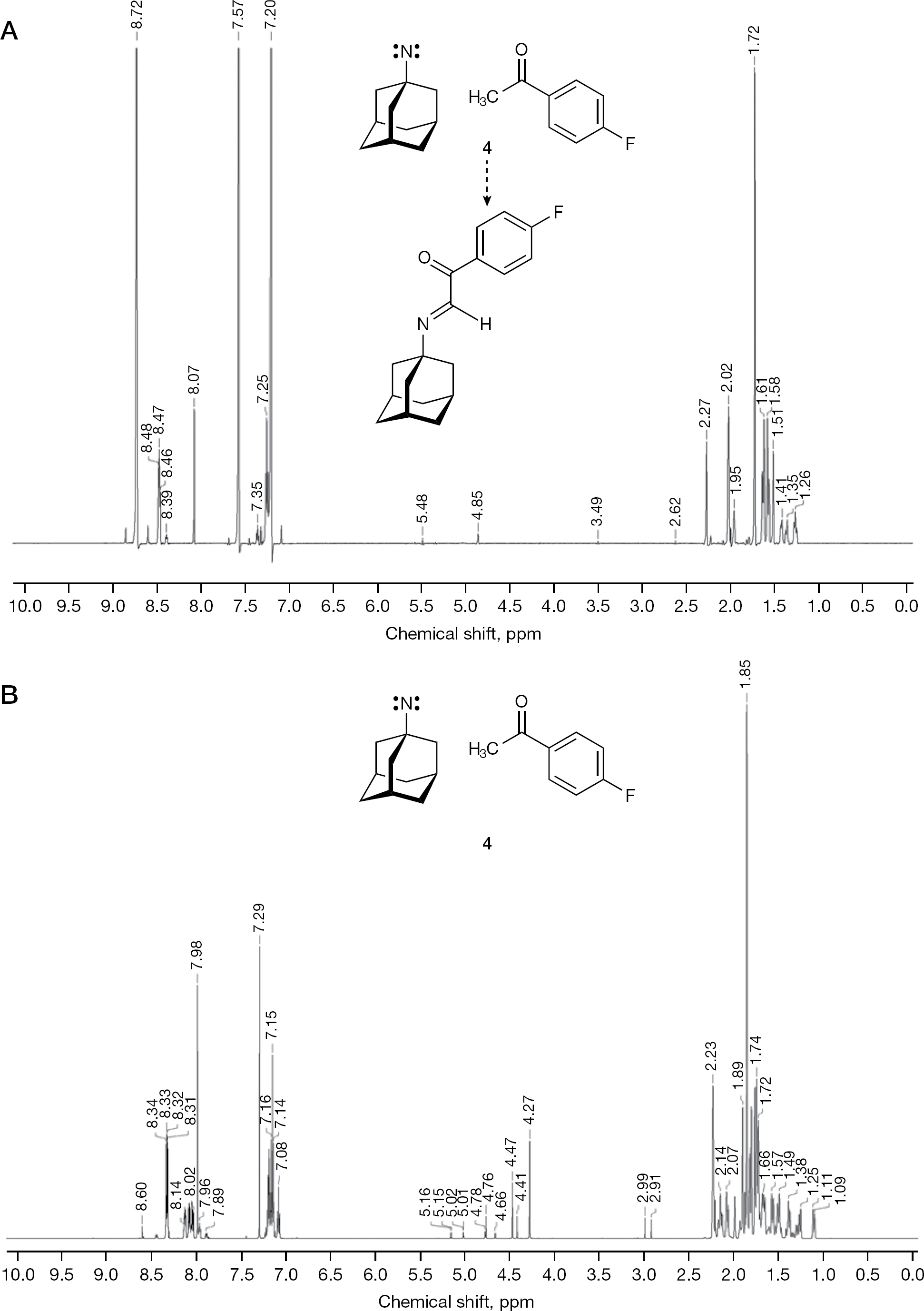
The proton nuclear magnetic resonance (1H NMR) spectra of 4 dissolved in [D5]pyridine and CDCl3
The 1H NMR spectra of 3 and 6 in CDCl3 were irregular (Additional file 1). They revealed signal diversification, which means that resonance signals for chemically equivalent atoms were split into a population of distinct, magnetically non-equivalent subpeaks. Additionally, proton signal area integrals were not integer and numerical calculable. The 13C DEPTQ NMR spectra of 3 and 6 in CDCl3 analogously revealed diversified 13C signals. However, 4 in [D5] pyridine gave NMR spectra (Figure 2A) with only few signal perturbations. The reason(s) for this signal perturbation(s) must stem from the presence of the singlet nitrene, but at present we cannot explain this phenomenon by certainly required in-depth physico-chemical investigations. It should be mentioned that repeated elemental analyses of 4 excluded the possibility that 4 incorporated N-hydroxy-1 or N-bromo-1 [34].
X-ray crystallographic investigation of 3
Proceeding further, 3 was crystallized from aqueous ethanol, and the obtained colourless needles were subjected to X-ray crystallographic analysis. One monoclinic crystal revealed an incommensurately modulated crystal phase [35–38]. The underlying average structure was solved by computational refinement and consists of a salt adduct (1:1) of 1 and 4-fluorobenzoic acid (3ox; Figures 3A–3C and Additional file 1). The formation of 3ox could be explained by the intrinsic impact of 1-adamantyl nitrene contained in 3 towards its ligand 4′-fluoroacetophenone. The possibility that 3ox exists preformed in 3 could be ruled out by analysis of the spectral data obtained with 3 and 4.
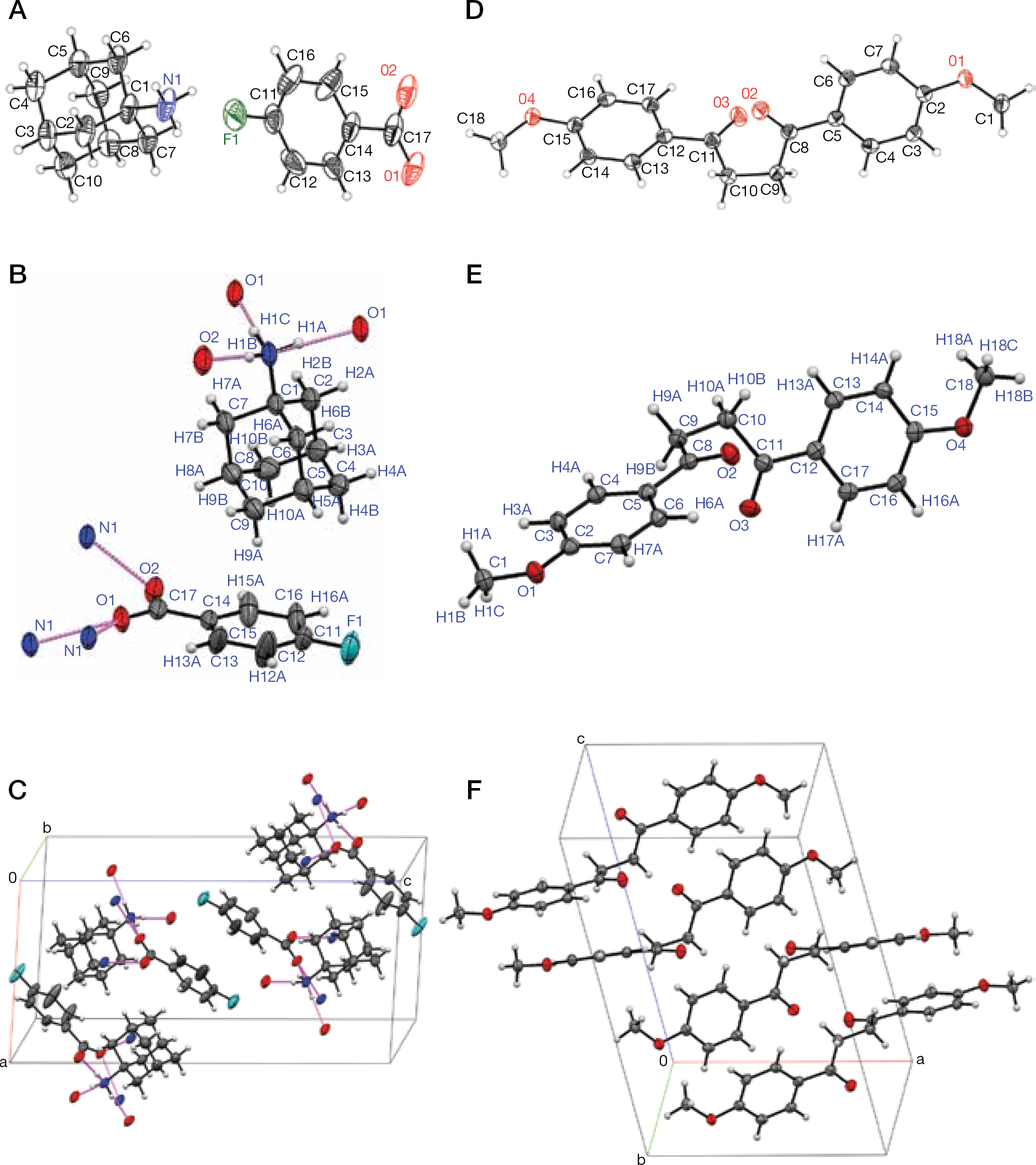
The X-ray crystallographic determined molecular structures of 3ox and 7
With this intriguing structure solved, the question arises as to why this nitrene in 3 is stable at all. Nitrenes (R–|N|) [39] are well-known as highly reactive intermediates in organic synthetic reactions, especially in rearrangements of Lossen-type (Lossen, Curtius, Schmidt, Hofmann) [40], and are short-lived and elusive [41]. Therefore, 3 was subjected to electron paramagnetic resonance spectroscopy (EPR); 3 dissolved in chloroform gave essentially no paramagnetic resonances at 293 K (data not shown). Since only singlet nitrenes, with all electrons paired, lack EPR signals at room temperature, 3 must represent a singlet nitrene (R–|N|). Triplet nitrenes (R–|N··) show strong paramagnetic resonance signals [42]. It is postulated that 3 and 6 are intrinsically stabilized by intermolecular carbonyl (C=O)–singlet nitrene nitrogen (1-adamantyl–|N|) interaction(s). The intramolecular version of this stabilization was proved to be the case in benzoyl singlet nitrene [(C6H5– =O)–|N|] by Gritsan et al. [43] and Pritchina et al. [44]. Therefore, 3 and 6 could be seen as intermolecular stabilization analogues of intramolecular-stabilized singlet nitrenes.
X-ray crystallographic investigation of 6
By analogy to 3, 6 was crystallized from aqueous ethanol. Colourless columns were obtained and subjected to X-ray crystallographic analysis. The crystal structure was solved and refined by standard methods (Figures 3D–3F and Additional file 1). It consisted of non-solvated, monoclinic 1,4-bis(4-methoxyphenyl)butane-1,4-dione (7), a molecule which is known since 1891 (Additional file 1) and then was occasionally re-synthesized [45]. The compound 6 obviously contained 7 as an adduct component which was created by intermolecular debromination of 5 [45].
Reaction of 6 with L-folic acid dihydrate
New insights into the chemical reactivity of 3 and 6 were sought. As a reaction partner of 6 the haematopoietic B vitamin L-folic acid dihydrate (8) was selected. After reaction of 6 with 8, a yellow hydrogel resulted, which was dehydrated to the yellow-orange powder 9. According to the 1H NMR and 13C DEPTQ NMR spectra (Additional file 1), the structure of 9 was deduced as solvated molecular adduct of 1,4-bis(4-hydroxyphenyl) butane-1,4-dione, 7 and a folate dimer in the molar ratio 0.2:1:1 (Figure 4). Obviously, the 1-adamantyl singlet nitrene contained in 6 attacked the methoxy group of its complexation partner 7 in α-position to yield an unknown reactive species, a formaldehyde-related C1 equivalent, which dimerized 8 at its secondary carboxamide nitrogen.
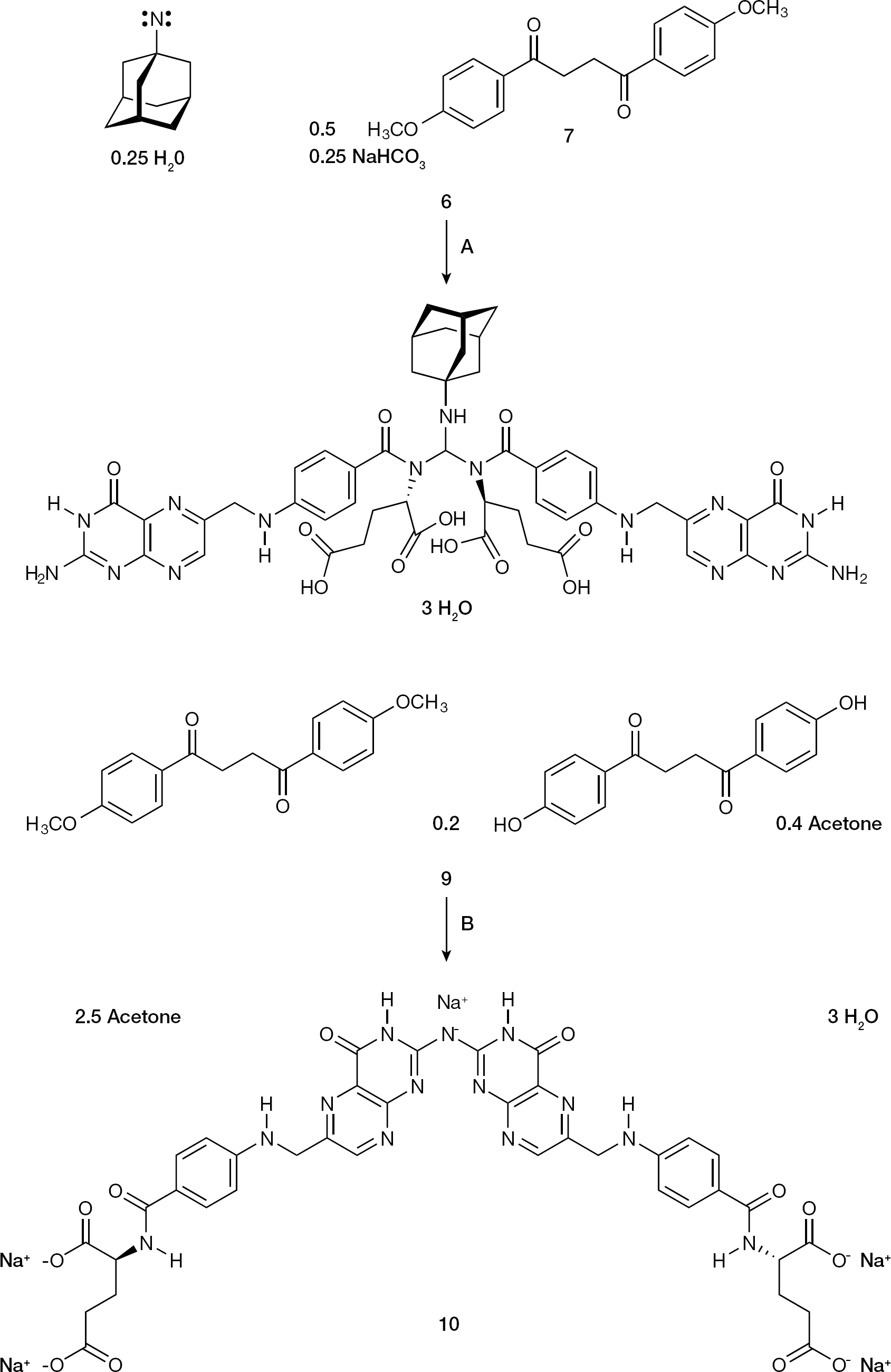
The chemical reactivity of the 1-adamantyl singlet nitrene-containing adduct 6 towards the haematopoietic B vitamin folic acid
Synthesis of the folate dimer 10
Compound 9 was subjected to NaHCO3-supported hydrolysis (pH 8) in aqueous acetone (Figure 4). The evolving product 10 was precipitated with acetone. According to the 1H NMR and 13C DEPTQ NMR spectra of 10 in deuterium oxide (D2O; Additional file 1), the C1-linker was lost during hydrolysis. The spectra revealed close similarity, but not identity, to the NMR spectra [46,47] of disodium L-folate (that is, 8 in D2O/NaOD). A decision regarding whether 10 was identical to solvated disodium L-folate (11) could not be made. Therefore, disodium L-folate tetrahydrate (11) was synthesized from 8. Additionally, the NMR spectra of 8 dissolved in deuterated DMSO ([D6]DMSO) were recorded (Additional file 1). The employed commercial 8 was not entirely pure and contained 1.5% (n/n) of an equimolar mixture of N-(4-aminobenzoyl)-L-glutamic acid and pterin-6-carboxaldehyde.
We confirmed that 10 and 11 were not identical, and that 10 represented another folate dimer, dimerized at the 2-amino group in the pterin part (Figure 4). For dimerization ammonia (NH3) must have split off from two molecules of 11, a phenomenon which was recently observed by one of us during studies with vitamin A-derived (retinoid) thiosemicarbazones [48]. The comparison of optical rotations of 8, 9, 10 and 11 was informative. At the concentration of 0.1% (w/v) 8 showed an [α]D20 of 14.0° (in DMSO), 9 of 23.0° (in DMSO), 10 of 18.0° (in water) and 11 of 49.0° (in water). Folate optical activity was retained in the syntheses of 9, 10 and 11, and the non-identity of 10 and 11 could be further substantiated. By comparison, the 1H NMR spectra of 8 and 11, as well as the 13C DEPTQ NMR spectra of 8 and 11 (Additional file 1), finally secured the NMR assignments.
Synthesis of 12 from 3 by C–H insertion
It is possible that 3 could react with the nucleoside cytidine. When 3 was refluxed with cytidine in acidified aqueous acetone, the product 12 was isolated by crystallization (Figure 5). According to its 1H NMR, 13C DEPTQ NMR and FT–IR spectra (Additional file 1), 12 represents the initially sought N-(4-fluorophenacyl)-1 as solvated hydrochloride (Figure 5). The weak base cytidine (pKa 4.22 at 298 K) [49] was inert towards 3 per se, but mediated transfer of 4′-fluoroacetophenone to the 1-adamantyl nitrene nitrogen, by mediating C–H insertion, a reaction typical for nitrenes [40].
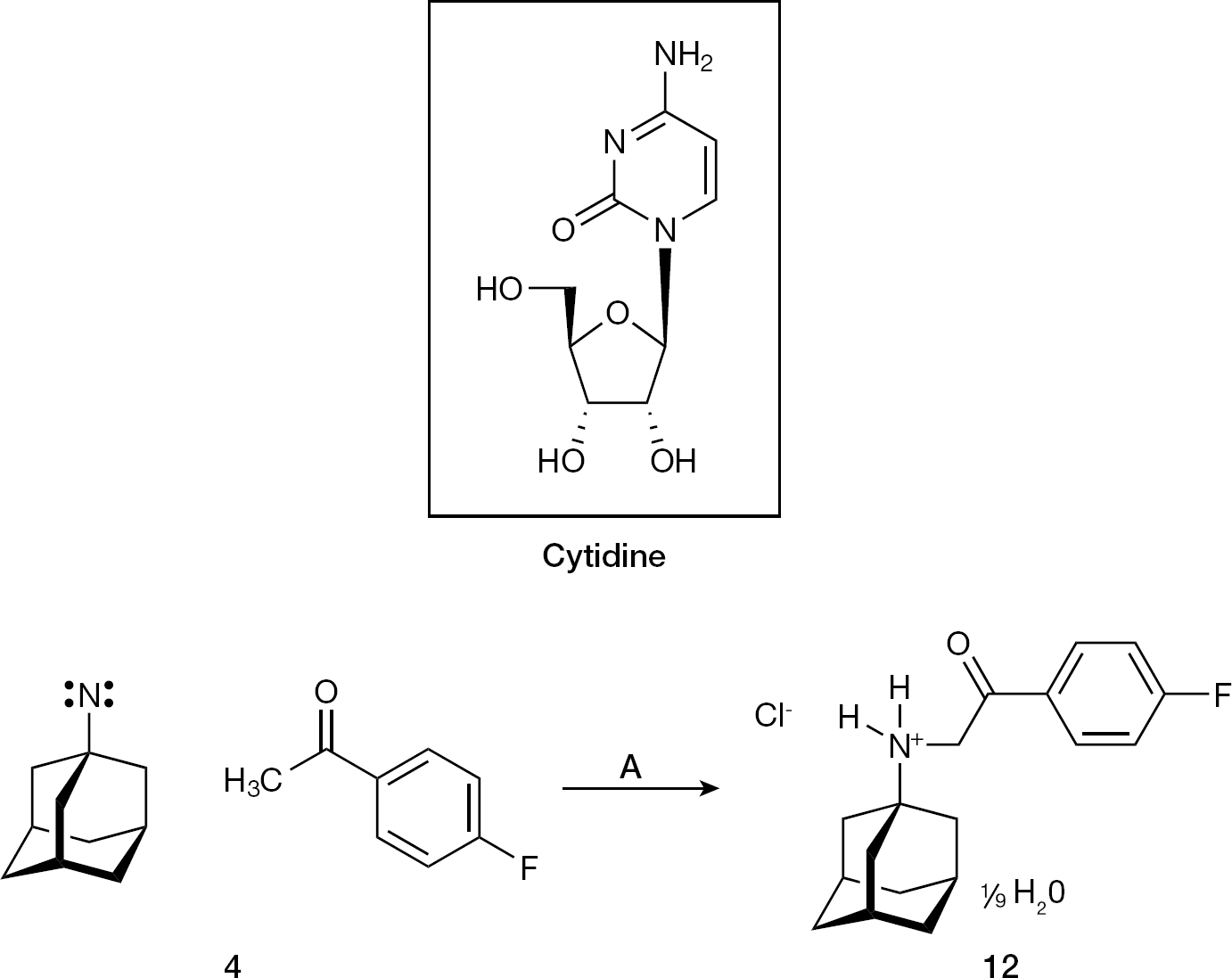
The chemical reactivity of the 1-adamantyl singlet nitrene-containing adduct 4 towards the nucleoside cytidine
Anti-HIV-1 activities of 1 xHCl, 4, 8, 9, 10, 11 and 12
The investigation on potential anti-HIV-1 activity was performed with most of the synthesized compounds, as well as with a suitable reference control. The in vitro anti-HIV-1LAI [15] activity of compounds 1 xHCl, 4, 8, 9, 10, 11 and 12 in human PBMC was determined (Table 1). 1 xHCl, 4, 8, 10, 11 and 12 did not show any anti-HIV-1 activity. However 8, in contrast to its disodium salt 11, exhibited toxicity in PBMC, indicating that the acidic nature of 8 may exert pH-related effects on human lymphocyte cells. Compound 9 was an effective inhibitor of HIV-1LAI in PBMC, but its antiretroviral activity was diminished by a freezing–(re-thawing) cycle (FRC) of the employed DMSO stock solution in which 9 was dissolved. After first FRC, activity was seriously affected, and, after second FRC, activity was completely lost. This strongly points to hydrolytic instability of the modified folate in 9, and proves that the antiretroviral activity is exerted by this folate analogue. The other components in 9 are chemically stable under the FRC conditions applied to the DMSO stock solution of 9. Evidence for the hydrolytic instability of 9 comes from literature data describing the reactions of N,N′,N″-tris(acyl) methanetriamines [50] and N,N′-[(dimethylamino) methylene]bis(succinimide) [51]. Because 9 represents a substituted N,N′-[(1-adamantylamino)methylene] bis(benzamide), hydrolytic instability towards the absorbed water content in the hygroscopic DMSO stock solution of 9 is to be expected. This is a consequence of the high steric tension created in 9 by the bulky-substituted methine carbon of the triaminomethane [H–C(NH2)3] type.
Anti-HIV-1LAI activities of 1 xHCl, 4, 8, 9, 10, 11 and 12 in primary human peripheral blood mononuclear cells
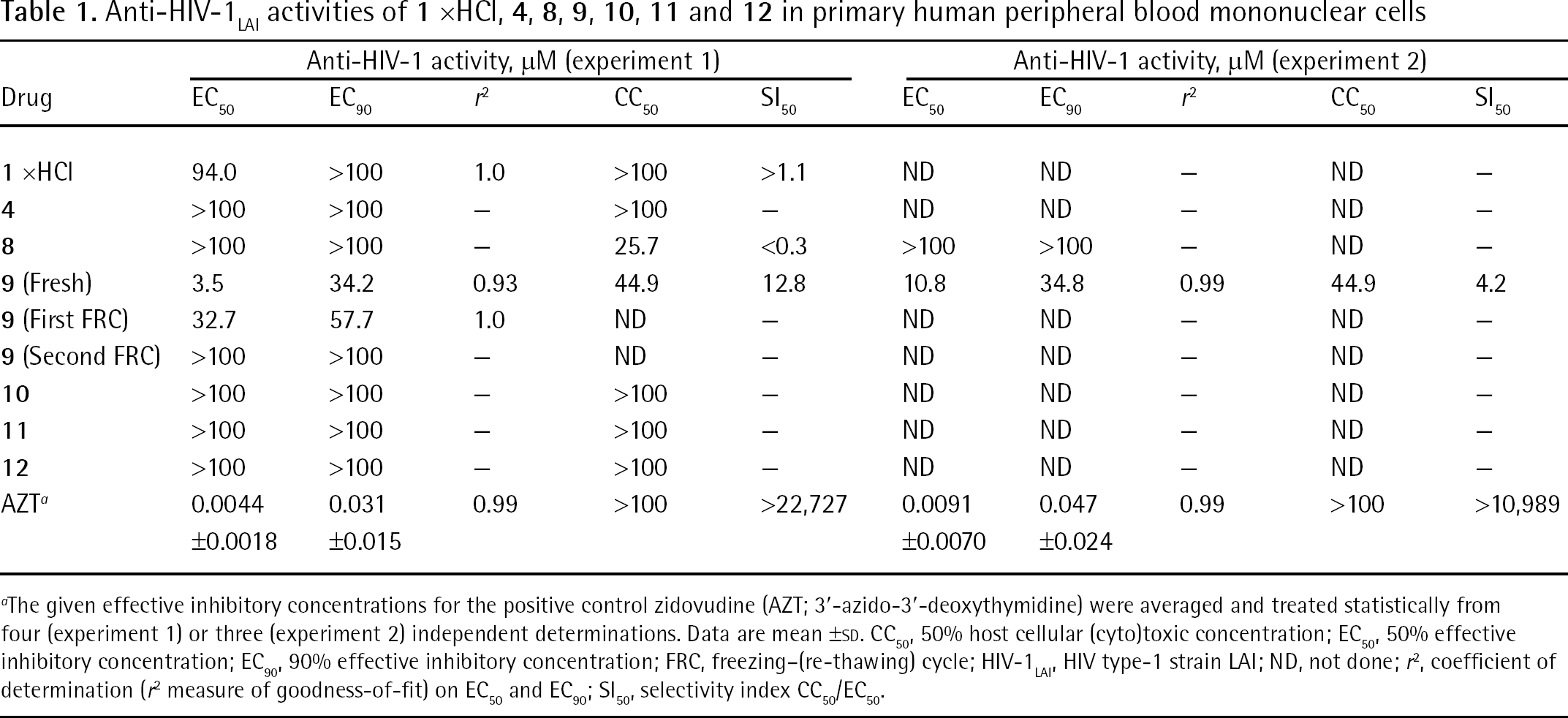
The given effective inhibitory concentrations for the positive control zidovudine (AZT; 3′-azido-3′-deoxythymidine) were averaged and treated statistically from four (experiment 1) or three (experiment 2) independent determinations. Data are mean ±sd. CC50, 50% host cellular (cyto)toxic concentration; EC50, 50% effective inhibitory concentration; EC90, 90% effective inhibitory concentration; FRC, freezing–(re-thawing) cycle; HIV-1LAI, HIV type-1 strain LAI; ND, not done; r2, coefficient of determination (r2 measure of goodness-of-fit) on EC50 and EC90; SI50, selectivity index CC50/EC50.
Through the comparison of the activity of 9 and the inactivity of 10, first hints for the mechanism-of-action of 9 can be deduced. Both 9 and 10 are tetracarboxylates, completely dissociated at physiological pH 7.4 (for folates: pKa [α-carboxylic acid] 3.3–3.5, pKa [γ-carboxylic acid] 4.6–4.8 [52]). The difference between the folate analogue cores of 9 and 10 is the distance between their two pairs of carboxylates. In 9 they are arranged in close proximity (α-carboxylates 5–6 Å; γ-carboxylates 12–13 Å), whereas in 10 they are remote to each other (α-carboxylates 9–10 Å; γ-carboxylates 18–19 Å; Figure 4 and Additional file 1). These structural characteristics are reminiscent of polyanionic inhibitors of HIV-1 external envelope glycoprotein 120 kDa (gp120) third variable region (V3 loop), notably the tetrasulfonate FP–21399 [53] and the hexasulfonate suramin (Additional file 1). We propose that 9 binds to HIV-1 gp120 V3 loop analogously to FP–21399 and other polyanionic drugs, and that this interaction builds the major basis for the observed HIV-1-suppressing effect of 9.
Activity of 12 against selected respiratory viruses
Because of the close chemical relationship to 1 xHCl, 12 was tested for inhibitory action on selected respiratory viruses (Table 2). The effects of 12 on the replication of the influenza viruses A/California/07/2009 (pandemic swine-origin H1N1, amantadine-resistant due to S31N mutation in M2 protein), A/Perth/16/2009 (seasonal H3N2, amantadine-resistant due to S31N mutation in M2 protein), A/duck/Minnesota/1525/81 (low pathogenic avian influenza H5N1, amantadine-susceptible), A/Vietnam/1203/2004 (highly pathogenic avian influenza H5N1, amantadine-resistant due to L26I and S31N mutations in M2 protein), and B/Florida/4/2006 (naturally amantadine-resistant) were determined. Additionally, 12 was tested for inhibitory action on human parainfluenza virus type 3 and SARS coronavirus replication. Compound 12 was inactive when evaluated for inhibition of influenza A/California/07/2009, A/Perth/16/2009, A/Vietnam/1203/2004 and B/Florida/4/2006 virus replication, and also did not inhibit human parainfluenza virus type 3 and SARS coronavirus replication. By contrast, 12 did inhibit the replication of amantadine-susceptible avian influenza A/duck/Minnesota/1525/81 (H5N1) virus. Since 12 inhibited only the amantadine-susceptible virus, it could be concluded that 12 acts like a true analogue of 1 xHCl with retained N-basicity. For the low pathogenic H5N1 virus, the EC50 of 1 xHCl was determined to be 0.54 μg/ml (2.88 μM) [22], whereas the corresponding EC50 of 12 was found to be 0.95 μg/ml (2.92 μM). By contrast with compound 9, 12 was not sensitive to hydrolysis, and the hygroscopic DMSO stock solution of 12 did not lose activity during multiple FRCs. The substituted phenacylamine 12 is reasonably stable to hydrolysis, as was indicated by its synthesis method. The compounds 4, 8, 9, 10 and 11 were not tested against respiratory viruses, since folates were observed to either promote influenza virus replication (42 μM 8 added to cell culture medium efficaciously enhanced influenza A virus haemagglutinin titres [54]), or because of potential toxicity (that is, 4 is precluded from being used as a therapeutic intervention in vivo because of its intrinsic chemical reactivity).
Anti-respiratory virus activities of 12

The reference drug used was ribavirin (1-β-D-ribofuranosyl-1H-1,2,4-triazole-3-carboxamide), except for severe acute respiratory syndrome (SARS) coronavirus, where the reference drug EP128533 (Maxim Pharmaceuticals, San Diego, CA, USA) [25] was applied. For influenza A/duck/MN/1525/81 virus the reference drug oseltamivir carboxylate (GS4071) [(3R,4R,5S)-4-(acetylamino)-5-amino-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid] was included in the neutral red assay (mean ±sd 50% effective inhibitory concentration [EC50] 0.061 ±0.053 μg/ml; 50% host cellular [cyto]toxic concentration [CC50]>10 μg/ml). b Here the 90% effective inhibitory concentration (EC90) was determined. CCID50, cell culture infective dose required to infect 50% of host cells per well; MA-104, embryonic African green monkey kidney epithelial cells; MDCK, Madin–Darby canine kidney cells; Vero 76, African green monkey kidney epithelial cells; Neutral red, test for lysosomal uptake of 3-amino-7-(dimethylamino)-2-methylphenazine hydrochloride (neutral red vital stain); SI50, selectivity index CC50/EC50; VYR, virus yield reduction assay.
Discussion
The experimentally observed chemical reactivity of 4 could be interpreted over the intermediate formation of three-membered rings (Figure 6). During crystallization of 4 (contained in 3) for X-ray crystallography the 1-adamantyl singlet nitrene underwent addition to the carbonyl group of 4′-fluoroacetophenone to yield an oxaziridine. The oxaziridine rearranged to an imidoester which was hydrolyzed to the strong base 1 (pKa 10.71 at 293 K [55]) and 4-fluorobenzoic acid. In the synthesis of 12, 1-adamantyl nitrene added to the enol form of 4′-fluoroacetophenone to yield an aziridin-2-ol which equilibrated to the free base of 12. It could be unequivocally excluded that the products 3 and 4 contain an arylglyoxal alkylimine in situ (Figure 6). By contrast, the NMR spectra of 4 in [D5]pyridine (Figure 2A and Additional file 1) revealed the presence of (2E)-2-(1-adamantylimino)-1-(4-fluorophenyl) ethanone, an arylglyoxal alkylimine [56,57,58]. Its formation must be induced by [D5]pyridine, since 4 dissolved in CDCl3 gave spectra (Additional file 1) typical for 1-adamantyl nitrene-containing materials.

The interpretation of the experimental reactivity of the 1-adamantyl singlet nitrene-containing adduct 4
Our results could contribute to the development of new amantadine-derived anti(retro)viral drugs by utilization of the stabilized 1-adamantyl nitrene complexes. It is expected that multiple (bio)chemical reaction partners for the stabilized 1-adamantyl nitrene complexes will be identified in the future, leading to unprecedented new arrays of molecular entities which will enable successful application in anti(retro)viral chemotherapy.
Footnotes
Acknowledgements
We thank O Burghaus (Philipps University of Marburg, Marburg, Germany) for conducting the EPR measurement on 3, and J Schweer, S Müller, C Freudenberger, T Westfeld, E-M May, D Wiegel, W Wuebbolt and R Sachs for analytical services (Currenta GmbH & Co. OHG). This work was supported in part by NIH grant 2P30–AI–050409 (Center for AIDS Research), and by the Department of Veterans Affairs (to RFS). In vitro work with respiratory viruses was partially supported by NIH/NIAID contract numbers HHSN272201100019I/HHSN27200001/B01 and HHSN272201100019I/HHSN27200002/B05. We thank S Orwin for technical work with in vitro assays. We are obliged to K Hecker (HEKAtech GmbH) for expert elemental analyses.
Atomic coordinates are deposited in The Cambridge Crystallographic Data Centre under accession numbers CCDC 872118 (3ox) and CCDC 826749 (7). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
The authors declare no competing interests.