
Abstract
Since its approval for clinical use in 2001, tenofovir (TFV) has become one of the most frequently prescribed nucleotide analogues used in combination with other antiretroviral agents against HIV-1 infection. Although reverse transcriptase inhibitors (RTIs) including TFV have been shown to be highly potent with reasonable safety profiles in the clinic, drug resistance hinders the effectiveness of current therapies and even causes treatment failure. Therefore, understanding the resistance mechanisms of RT and exploring the potential antiviral synergy between the different RTIs in combination therapies against the resistance mechanisms would greatly improve the long-term efficacy of existing and future regimens. We have studied the pyrophosphorolytic removal of TFV, a major resistance mechanism that RT utilizes, from two different viral sequences and observed interesting outcomes associated with the sequence context. Furthermore, addition of efavirenz, a non-nucleoside RTI, inhibits this removal process confirming the synergistic antiviral effects. This article highlights our recently published work on the viral sequence context contributing to the study of anti-HIV drug resistance in conjunction with the benefits of combining various RTIs that may have been neglected previously.
Introduction
Highly active antiretroviral therapy (HAART) also known as combination antiretroviral therapy (cART) is considered to be the most effective treatment in slowing the progression of HIV-1 infection and delaying the emergence of resistant mutants; however, it is incapable of eliminating HIV-1 infection [1]. There are several different stages of the HIV lifecycle that are targeted with major efforts centred around HIV reverse transcriptase (RT), HIV protease and more recently viral entry, attachment and integration [2]. Among all the developed anti-HIV agents, the drugs targeting HIV-1 RT continue to be the foundation of cART, and are divided into two classes. Firstly, nucleoside/nucleotide RT inhibitors (NRTI/NtRTIs; NRTI and NtRTI are interchangeably used and indicated as N(t)RTI throughout the text) are prodrugs that require intracellular conversion into the pharmacologically active triphosphate/diphosphate forms and exert their antiviral activities via chain termination due to the lack of a 3′-OH group after being incorporated into the growing viral DNA strand (reviewed in [3]). Secondly, non-nucleoside RT inhibitors (NNRTIs) possess diverse structures and do not require any cellular activation for blocking HIV replication. These inhibitors bind to an allosteric hydrophobic pocket 10 Å away from the RT polymerase catalytic site, causing long-range distortions in the catalytic site, thus disturbing the incorporation of natural substrates (reviewed in [4,5]).
The latest consensus is to combine at least three drugs from two different classes to circumvent or diminish the emergence of resistant HIV-1 strains. Although cART effectively controls the viral load, the therapy could lead to failure following the appearance of drug-resistant virus. Because RT lacks a proofreading mechanism, the errors that arise during each viral lifecycle result in rapid emergence of antiretroviral drug resistance [6]. Therefore, it is important to understand the resistance mechanisms and potential drug interactions in order to develop more effective strategies for treating HIV infection. Previous studies utilizing the various combinations of N(t)RTIs and NNRTIs showed antiviral synergistic effects for the inhibition of viral replication in cell culture [7–9] and in a clinical setting [10,11]. Tenofovir (TFV), the active drug of tenofovir disoproxil fumarate (TDF) prodrug, is the only approved N(t)RTI for clinical use in HIV treatment and one of the most effective and frequently prescribed RTIs (Figure 1). TFV is used in several co-formulations that are administered as once-daily single tablet regimens, such as Truvada® (consisting of TDF and emtricitabine [FTC] as another NRTI), Atripla® (consisting of TDF and FTC, and efavirenz [EFV] as an NNRTI; Figure 1) and Complera® (consisting of TDF and FTC, and rilpivirine as an NNRTI). Recently, FDA approved Stribild®, which is the co-formulation of four compounds including TDF and FTC as N(t) RTIs, elvitegravir as the integrase inhibitor and cobicistat as a boosting agent. We have been studying the mechanism of antiviral synergistic effects between the components of Atripla®, which is considered to be the gold-standard for the first-line therapy [12,13]. This article summarizes our recent findings on a comparative study of TFV excision from two different primer-template sequences derived from the HIV-1 genome [13] in relation to earlier work by others, with a particular emphasis on the additional effects of EFV during this resistance process.

Components of Atripla® co-formulation
Resistance mechanisms against tenofovir
TFV has an acyclic moiety instead of a deoxyribose sugar ring that links the adenine base to a phosphonate group and replaces the α-phosphate portion of the triphosphate form of the nucleotides. Therefore unlike NRTIs, TFV requires only two consequent phosphorylations to establish the active diphosphate form (TFV-DP) in order to be incorporated as a chain terminator into the DNA. Moreover, this phosphonate group is resilient to excision by 3′-5′exonucleases which makes it more potent against the virus. However, HIV-1 exploits two distinct resistance mechanisms against any N(t)RTIs, including TFV. These mechanisms have been emphasized in several reviews [14,15]. First, selection of one or more resistance mutations in RT leads to an increase in discrimination against N(t)RTIs by reducing the enzyme's binding affinity to the nucleoside analogue triphosphate derivatives or decreasing the incorporation rates of these nucleotides. For instance, emergence of the K65R mutation in RT reduces susceptibility to TFV and discriminates against TFV by decreasing the rate of nucleotide incorporation (kpol) for TFV-DP compared to the wild-type RT [16–20]. The other mechanism involves the phosphorolytic removal (representing the reversal of the polymerization reaction) of the chain-terminating N(t)RTI from the primer terminus subsequent to its incorporation into viral DNA [21,22]. In this resistance mechanism, RT utilizes ATP or inorganic pyrophosphate (PPi) as the pyrophosphate donor to remove the incorporated NRTI-monophosphate or N(t)RTI such as TFV, thus restoring the DNA synthesis (illustrated in Figure 2A and 2B). In most cases, the physiologically relevant pyrophosphate donor is cellular ATP, where it nucleophilically attacks the 3′-end of a chain-terminated DNA primer and yields a dinucleoside tetraphosphate, while generating a free 3′-OH at the primer terminus. Tu et al. [23] showed the binding interactions of wild-type RT and zidovudine (AZT)-resistant RT structures bound to the covalently attached 5′pAZTpppA (dinucleoside tetraphosphate), in which mutations affect the ATP binding site and possible interactions with the residues surrounding the site.
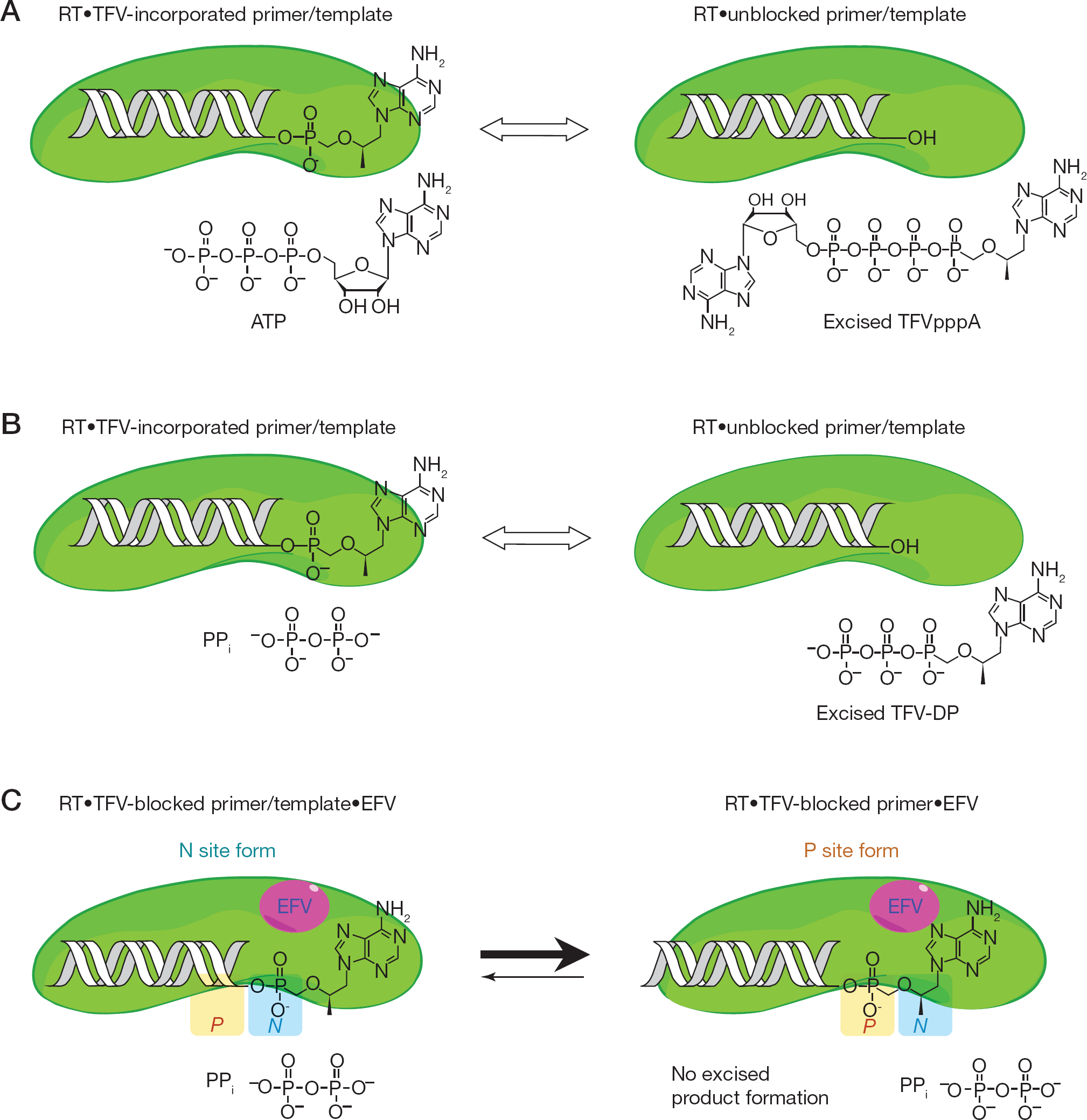
Illustration of TFV excision reaction
The role of sequence context in studying N(t)RTI resistance
Previous biochemical studies on the phosphorolytic removal efficiency of TFV reported contradictory results, which could possibly be due to using different primer-template sequences in each experiment [24–27]. Although the effects of primer-template sequence on ATP-mediated N(t)RTI removal has been explored through substitution within the same sequence space [28], there are no comparative reports with different HIV-1-specific sequences until our recent excision study using both polypurine tract (PPT) and primer binding site (PBS) sequences. In order to verify our hypothesis, we selected these essential PPT and PBS viral sequences as primer-templates to explore the sequence dependency of TFV excision efficiency catalysed by RT. The importance of PBS and PPT sequences for the HIV-1 replication is explained comprehensively in a review [4]. Briefly, the PBS sequence within the viral RNA genome anneals to a cellular tRNA that is used to prime the synthesis of minus-strand viral DNA. The RNase H activity of RT cleaves the genomic RNA template at various points generating short RNA pieces during minus-strand DNA synthesis. Among these formed RNA fragments, the PPT sequence serves as a primer to initiate the plus-strand DNA synthesis, which results in the double-stranded DNA viral genome.
Our analysis of ATP-mediated TFV removal from PPT and PBS primer-template sequences showed an adenosine monophosphate (AMP)-incorporated primer band rather than TFV-excised product band (n-1) formation [13]. Furthermore, TFV excision is eightfold more efficient with PPT sequence over PBS sequence derived oligonucleotides. We believe that RT not only utilizes ATP to remove TFV from both primers, but also incorporates a ribonucleotide as a substrate to the unblocked DNA primers since AMP and TFV possess the same nucleobase moiety. This observation provides another example of the low fidelity of HIV-1 RT, which is an evolutionary trait required to maintain high viral mutation rates. PPi-mediated TFV removal also showed a similar trend of more efficient removal with PPT primer/template. Consequently, we hypothesize that the unique sequence motifs of PPT, including two (A)4-tracts followed by one (G)6-tract at the 3′-terminus of the sequence, could be the reason for enhanced excision properties compared to the PBS sequence. Previous investigations have shown that A-tract and G-tract DNA sequences have distinctive helical structures with characteristic minor groove distances compared to the canonical DNA sequences [29–32]. These exceptional structural features of PPT sequences could modulate the binding conformation and binding specificities of any protein to these sequences [33–35]. Moreover, viral RTs from HIV-1, Moloney murine leukemia virus and Rous sarcoma virus utilize the aforementioned structural elements of PPT to make specific interactions for preventing its degradation by the RNase H activity and maintaining the precise RNase H cleavage in the genome sequence for plus-strand priming [36–39]. Several laboratories have solved the crystal structures of HIV-1 RT in complex with the DNA:RNA hybrid PPT duplex [40–42] and also showed that the substitutions at the 5′-end of HIV-1 PPT sequence, 3′ (G)6-tract portion along with the randomization of the entire PPT sequence could lead to low infectivity [43–45]. Additionally, the combination of mutations in the PPT sequence along with mutations in HIV-1 RT disturbed the overall structure of the complex and showed strong effects on the specificity and the rate of PPT cleavage [46]. These results point to the evolutionary pressure to preserve the intact PPT sequence due to its required sequence-based structure for correct cleavage and viral fitness. Therefore, HIV-1 RT excises TFV from the PPT region more efficiently than the PBS sequence.
The role of efavirenz on tenofovir removal
A number of studies have elucidated mechanisms of synergy between N(t)RTIs and NNRTIs at the enzyme level. The bound NNRTI reduces both the rate of excision reaction and affinity of ATP to RT [47,48]. Most recently, Feng et al. [49] reported that formation of NNRTI-mediated stable complex prolongs the chain-termination effects of N(t)RTIs. Also, NNRTI binding diminishes N(t)RTI excision via disturbing the rate and pattern of RNase H cleavage of RT [50–52]. According to our recent study, EFV binding to the allosteric site of RT could facilitate small changes within the polymerase active site and/or the nucleotide acceptor molecule (ATP or PPi) binding site of the RT·TFV-terminated DNA complex [13]. These long-range interactions upon NNRTI binding lead to an equilibrium shift towards the priming site (P-site) from the nucleotide-binding site (N-site), thus generating a non-productive binding orientation for TFV removal (Figure 2C). As a result, EFV forms a stable complex that is mechanistically very similar to the nucleotide-induced dead-end-complex formation but only differs in lacking the presence of the next complementary nucleotide at the N-site [53,54].
Conclusions and future directions
The mechanisms of resistance have been studied for two decades; however, deciphering the new molecular mechanisms underlying HIV persistence continues to be crucial as more antiviral drugs are becoming available in the clinic with different mode of actions. Further biochemical studies using different chain terminators with PPT, PBS and other viral sequences will particularly enlighten the sequence-dependent N(t)RTI removal efficiency. PMPG ((R)-9-[2-(phosphonomethoxy)propyl]guanine), the guanine derivative of TFV, could serve as a suitable candidate to explore the excision efficiency profile with both sequences since the only difference is the nucleobase in both nucleotides. Additionally, crystallographic analysis of HIV-1 RT in complex with various chain-terminated primer-template sequences might provide more insight by capturing the steps of the excision process with structural evidence. In conclusion, the presented data in this article accentuates the importance of sequence scaffold for assessing resistance profiles of newly developed drugs.
Combination therapy helps tackle the drug-resistance problem up to a certain extent via antiviral synergy, yet there is still much room for improvement. We propose that EFV could induce just enough conformational changes in RT to change the whole cascade of the TFV excision reaction. Ultimately, these assumptions should also be tested using various NNRTIs to generalize their possible effects on N(t)RTI resistance.
Footnotes
The authors declare no competing interests.