
Abstract
Background
For patients with HBV infection who have decompensated cirrhosis (DC), a higher dose (1.0 mg/day) of entecavir is recommended than that used for those with compensated disease (0.5 mg/day), though with very little supporting data. We therefore compared the viral suppression achieved with 0.5 mg/day and 1.0 mg/day of entecavir in patients with HBV-related DC (NCT03345498).
Methods
Treatment-naive patients with HBV-related DC and serum HBV DNA titre exceeding 100,000 IU/ml received either dose of entecavir for 24 weeks. HBV DNA concentration was measured in blood specimens collected at baseline and after 2, 4, 8, 12 and 24 weeks of entecavir treatment.
Results
Participants in the 0.5 mg/day (n=13) and 1.0 mg/day (n=16) groups had similar baseline hepatitis B e antigen (HBeAg) positivity rates (12/13 and 12/16; P=0.34) and median (range) log10 serum HBV DNA levels (6.81 [5.01–8.12] and 7.45 [5.24–8.65]; P=0.17). The two doses led to similar reductions in serum HBV DNA levels after 2, 4, 8, 12 and 24 weeks of entecavir administration. At 24 weeks, 3 of the 13 patients receiving 0.5 mg/day and 1 of the 16 patients receiving 1.0 mg/day of entecavir had undetectable serum HBV DNA. Serum albumin level showed significant and similar improvement at the end of 24 weeks in the two groups.
Conclusions
Treatment-naive patients with HBV-related DC can be treated with entecavir in a 0.5 mg/day dose instead of the higher 1.0 mg/day dose, without compromising the degree of virological suppression. ClincialTrials.gov number NCT03345498.
Introduction
Chronic infection with HBV, if untreated, can progress to cirrhosis. Cirrhosis is associated with development of portal hypertension, impaired liver function or both [1], leading finally to decompensated cirrhosis (DC), a clinical state characterized by the presence of serious complications, such as ascites, hepatic encephalopathy, variceal bleeding or jaundice. HBV infection is treated with nucleoside/nucleotide analogues (NAs), such as entecavir or tenofovir, which inhibit viral replication [2,3]. In patients with HBV-related DC, entecavir is preferred over tenofovir, because long-term use of the latter can result in impairment of bone density and of renal function [4].
In patients with HBV infection, entecavir is used in a dose of 0.5 mg/day. However, for patients with decompensated cirrhosis, a higher dose, that is, 1.0 mg/day, is recommended by all the major international professional associations for the study of liver diseases [2,3,5]. However, hardly any data are available to support this recommendation. The use of higher dose of entecavir poses a disadvantage of increased cost of treatment, particularly in low-income countries. In fact, some physicians in such parts of the world treat the patients who cannot afford a higher dose with the usual 0.5 mg dose of entecavir. Hence, there is a clear need to compare the 0.5 mg/day and 1.0 mg/day doses of entecavir in treatment-naive patients with HBV-related DC.
We undertook this study to compare the degree and time course of viral suppression through the first 24 weeks of treatment with 0.5 mg/day or 1.0 mg/day dosage schedules of entecavir in treatment-naive patients with HBV-related DC.
Methods
This prospective, two-group, open-label, observational study was conducted between January 2017 and December 2018 (NCT03345498). Patients with HBV-related DC, planned for entecavir treatment in a dose of either 0.5 mg/day or 1.0 mg/day by the treating physician, were enrolled after obtaining written informed consent. For inclusion, a patient had to have DC, serum HBV DNA titre >100,000 IU/ml irrespective of the hepatitis B e antigen (HBeAg) and anti-HBe test results, and be treatment-naive for HBV infection. Diagnosis of cirrhosis was based on a combination of clinical, biochemical, radiological and endoscopic findings. Hepatic decompensation was defined as per the criteria laid down by Asia-Pacific Association for the Study of Liver (APASL), that is, significant liver dysfunction as indicated by either serum bilirubin more than 2.5x the upper limit of normal and prolonged prothrombin time (prolonged by >3 s or international normalized ratio >1.5), or current or past occurrence of ascites or of hepatic encephalopathy [5].
Any patient with one or more of the following was excluded: prior treatment for HBV infection, namely an NA or pegylated interferon, clinical, biochemical or imaging evidence of hepatocellular carcinoma, coinfection with HCV or HIV, acute-on-chronic liver failure as per the criteria laid down by APASL [6], significant alcohol intake (exceeding 20 g/day for men and 10 g/day for women), another concomitant hepatobiliary disease, a complication that was expected to markedly limit survival duration (for example, haemodynamic instability, active sepsis, hepatorenal syndrome, etc), use of an immunosuppressive medication, portal vein thrombosis, or inability to return for the scheduled follow-up visits.
After obtaining written informed consent, relevant clinical and laboratory findings were recorded. Before starting entecavir, blood specimens were collected for HBV DNA measurement. Patients were followed up at 2, 4, 8, 12 and 24 weeks after the start of entecavir treatment. In each visit during the 24 weeks of treatment, compliance was recorded and reinforced. Any patient who had taken the assigned drug dose for ≥22 weeks during the 24-week follow-up duration was considered as compliant to treatment. At each visit, a blood specimen was collected for any tests for routine care, and for serum HBV DNA measurement. For the latter, serum was separated soon after blood collection and stored in aliquots at -80°C. All the HBV DNA assays were done in batches at the end of the study, using a real-time PCR assay (COBAS® AmpliPrep/COBAS® TaqMan® HBV Test v2.0; Roche, Basel, Switzerland) with a lower limit of quantification of 20 IU/ml. Biochemical liver function tests, and assays for HBeAg and anti-HBe (VIDAS®; Biomerieux, Marcy l'Etoile, France) were done at baseline and after 24 weeks of entecavir treatment.
A sample size of at least 13 patients per treatment group was needed to attain 90% power to demonstrate a difference of 1.0 log10 IU/ml in mean reduction of serum HBV DNA at week 24, assuming a mean reduction of 4.5 log10 IU/ml (with a standard deviation of 0.8) in the 1.0 mg/day treatment group [7], and a two-sided significance cutoff of 0.05.
HBV DNA titres were expressed as log10 IU/ml of serum. Categorical and continuous data were summarized as proportions and as median (range), respectively. The categorical and numerical data for the two treatment groups were compared using the χ 2 test and the Mann–Whitney U test, respectively. In particular, the decline in HBV DNA concentration at each time point from the pre-treatment baseline was compared between patients in the two groups using the Mann–Whitney U test. The study had been approved by our institution's Ethics Committee.
Results
Patients
A total of 44 patients with HBV-related DC were screened during the study period and 32 were enrolled in the study (Figure 1). Three of these patients, including two in the 1.0 mg/day entecavir group (after 4 and 12 weeks of treatment, respectively) and one in the 0.5 mg/day group (after 2 weeks) were lost to follow-up. We restricted our data analysis to the remaining 29 patients who completed the planned follow-up period of 24 weeks. All the 29 patients were judged as treatment compliant. Of these 29 patients, 13 received 0.5 mg/day and 16 received 1.0 mg/day of entecavir. The patients in the two groups were similar in clinical characteristics, laboratory parameters and liver disease severity scores (Table 1).
Baseline clinical and laboratory characteristics, and liver disease severity scores of study participants receiving the two different doses of entecavir

Categorical data and numerical data are expressed as proportions (%) and median (range) respectively; P-values refer to comparison of 0.5 mg/day versus 1.0 mg/day groups using Mann–Whitney U test. HBeAg, hepatitis B e antigen; INR, international normalized ratio; ULN, upper limit of normal.

Flow chart of study population
Change in HBV DNA with the two treatments over time
Median (range) pre-treatment serum log10 HBV DNA concentration in the 29 study subjects was 7.26 (5.01–8.65) IU/ml. After 24 weeks of entecavir treatment, only 4 (13.8%) of the 29 participants, including 3 of 13 receiving the 0.5 mg/day dosage and 1 of 16 receiving the 1.0 mg/day dosage, had achieved undetectable HBV DNA. The median (range) pre-treatment log10 serum HBV DNA level of these 4 patients (6.18 [5.01–7.39] IU/ml) was lower than that of the 25 patients who did not achieve undetectable HBV DNA at the end of 24-week treatment (7.43 [5.22–8.65] IU/ml), though the difference was not statistically significant (P=0.11).
Reductions in log10 serum HBV DNA level at weeks 2, 4, 8, 12 and 24 weeks of entecavir treatment, as compared to the pre-treatment level, in the 0.5 mg/day and 1.0 mg/day groups were comparable (Table 2 and Figure 2), as were the absolute serum HBV DNA levels at each time point between the two groups (Table 2). Five patients, who had detectable HBeAg before starting entecavir, became HBeAg-negative and developed anti-HBe antibody after 24 weeks of treatment. Three of those who cleared HBeAg had also achieved undetectable HBV DNA at 24 weeks.
Serum HBV DNA concentration (expressed as log10) and change from baseline at various time points in patients receiving different doses of entecavir

All data are shown as median (range). Data for decline in HBV DNA from baseline are presented as: (log10 [HBV DNA in IU/ml]before – log10 [HBV DNA in IU/ml]after). P-values refer to comparison of 0.5 mg/day versus 1.0 mg/day treatment groups using Mann–Whitney U test.
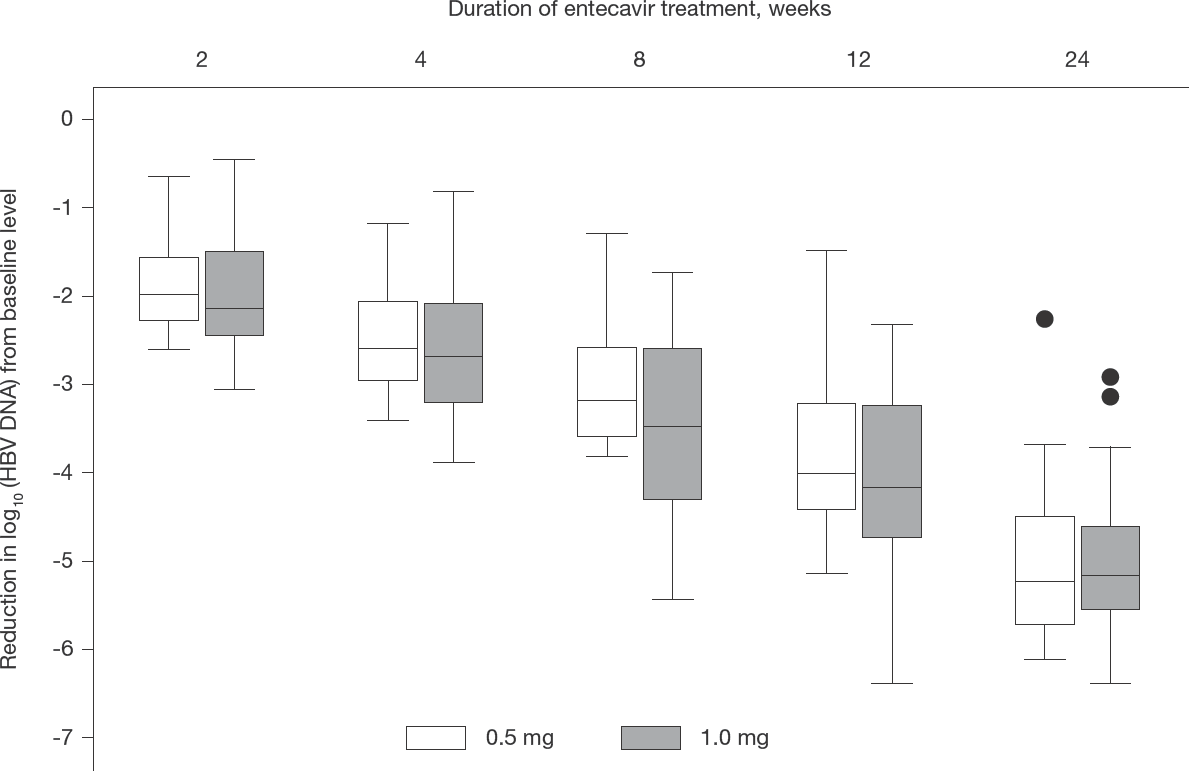
Comparison of sequential decline in serum HBV DNA levels (log10) from pre-treatment level between the two treatment groups
Effect of entecavir treatment on liver disease severity Serum albumin concentration showed a significant improvement after 24 weeks of entecavir treatment in both the 0.5 mg/day (0.7 [-0.4–1.7] g/dl; P<0.05) and 1.0 mg/day (0.4 [-0.7–1.9] g/dl; P<0.05) dosage groups. Overall, in the two groups taken together, the median (range) Child-Turcotte-Pugh score improved from 9 (6–12) to 8 (6–12), and the median MELD score changed from 16 (8–22) to 13 (7–28); however, these changes were not significant (P=0.09 and 0.13, respectively). The changes in laboratory measures of severity of liver disease (serum bilirubin, serum albumin and prothrombin time), serum creatinine, Child-Turcotte-Pugh score and MELD scores after 24 weeks of entecavir were comparable between the two treatment groups (Table 3).
Comparison of changes in laboratory parameters and liver disease severity scores after 24 weeks of entecavir treatment with the two different doses of entecavir

Data are expressed as median (range). CTP, Child–Turcotte–Pugh; MELD, model for end-stage liver disease.
Discussion
In this pilot study, we compared the virological suppression achieved in treatment-naive patients with HBV-related DC with the higher, that is, 1.0 mg/day, dosage recommended for patients with DC and the usual, that is, 0.5 mg/day, dosage of entecavir recommended for patients with compensated disease, using a cohort study design. The virological response to the two doses was found to be comparable at each of the various time points during the 24 weeks of treatment. The antiviral treatment was associated with an increase in serum albumin, though the absolute increase was small. Only a few patients (4 of 29; 13.8%) achieved undetectable HBV DNA after 24 weeks of entecavir treatment, and 5 developed HBeAg loss and HBeAg to anti-HBe seroconversion; for these outcomes, comparison between the two dosage schedules was not possible.
In the natural history of cirrhosis, appearance of decompensation marks a stage characterized by a high risk of serious complications and reduced survival. Regardless of the cause of cirrhosis, liver transplantation is the only definitive treatment for patients with DC. However, in patients with HBV-related DC, an effective antiviral drug can lead to marked and continued suppression of HBV replication, and significantly improved prognosis, including survival [8–10]. This has led to the recommendation to start an antiviral drug as soon as HBV-related DC is diagnosed, regardless of serum HBV DNA concentration. Of the several drugs that inhibit HBV replication, currently only those drugs that have a high barrier to development of antiviral drug resistance are recommended [2,3,5].
Entecavir is one such drug, with resistance to it occurring in only around 1% of those treated for as long as 7 years [11]. It is generally used in a dose of 0.5 mg/day in treatment-naive patients. A higher dose of 1.0 mg/ day is recommended for those who have previously failed treatment with lamivudine, the first NA used for the treatment of HBV infection [12], which had a low barrier to resistance. Thus, such patients had HBV with genomic variations that conferred drug resistance against lamivudine [13], and partial cross-resistance against entecavir. A similar phenomenon, though less marked, also occurs with other NAs, namely adefovir and telbivudine. This use of higher dose of entecavir in persons previously treated with lamivudine or telbivudine appears rational since it has been shown to achieve a better virological suppression than the 0.5 mg/day dose in such persons [12].
The higher dose of 1.0 mg/day has also been recommended for patients with HBV-related DC, regardless of their prior exposure to another NA, by several hepatology professional organizations [2,3,5,14]. However, documents that contain these recommendations do not provide any rationale for this increased dose. This empiric recommendation could be based on the following two rationales: potential to achieve a more rapid HBV suppression with a higher dose in these sicker patients, and to overcome any potential partial resistance to this drug because of prior use of a first-line NA with low barrier of resistance. However, the evidence to support this higher dose in patients with DC is quite limited and need further exploration.
Our data show a similar reduction in HBV DNA levels in patients with DC with the 0.5 mg/day and the 1.0 mg/day doses at all follow-up time points. This indicates that a more rapid or more marked reduction in HBV DNA cannot be the reason to justify administration of a higher entecavir dose in these patients. This argument is also supported by a previous study, in which Shim et al. [15] showed that median reduction in HBV DNA and the probability of achieving undetectable serum HBV DNA and HBeAg seroconversion, with the 0.5 mg/day of entecavir, was similar in patients with HBV-related DC and those with compensated HBV cirrhosis. In addition, in our study, the two doses of entecavir were associated with similar, albeit slight, improvements in serum albumin levels.
Furthermore, most of the patients with HBV-related DC who are started on entecavir are treatment-naive, and have not previously received another NA. Hence, they are unlikely to have a partial resistance to entecavir to justify a higher dose. Thus, we believe that a higher dose of entecavir is not justified in the patients with HBV-related DC who are being started on this drug. In fact, the use of the usual 0.5 mg/day dose would reduce the cost of HBV treatment to half – a major advantage since drug cost is a major barrier to access to HBV treatment worldwide [16]. The lower dose may also help reduce the risk of entecavir-induced myopathy [17], a particularly bothersome complication in patients with DC who often have a reduced muscle mass.
Our study had a few limitations, including an observational design, small sample size and a treatment duration of only 24 weeks which precluded assessment of differences in HBeAg or hepatitis B surface antigen (HBsAg) seroconversion, or rates of clinical complications or mortality between the two treatments. The non-randomized nature of the study might have biased the designation of the patients with more advanced liver disease into the 1.0 mg/day dose arm. This selection bias is refected by the higher international normalized ratio value and Child-Turcotte-Pugh score in 1.0 mg/day arm than 0.5 mg/day arm (Table 1). A double blind randomized controlled trial is needed for the definitive answer to this question. We were constrained to use the observational design because the common recommendation to use 1.0 mg/day dose of entecavir, albeit with very little evidence to support it, has become a de facto standard, making it difficult to obtain the permission from our Ethics Committee for a clinical trial with one group receiving a lower than the recommended dose of entecavir. Hence, we settled for an observational design, in the hope that if its results support our hypothesis, these may open the path for future larger interventional studies. Also, though our sample size may appear small, it was supported by our pre-hoc sample size calculation. In addition, though we could not assess the effect of different drug doses on clinical and other virological end points, we feel that these should eventually happen given that the two doses achieved comparable degrees of viral suppression.
In conclusion, our data show that treatment-naive patients with HBV-related DC treated with the usual 0.5 mg/day dosage of entecavir achieve a similar degree of viral suppression as that achieved with the double-dose regimen often recommended for such patients. Hence, such patients can potentially be treated with the lower dose of entecavir, with reduction in the cost of treatment and adverse events. At the very least, these data argue in favour of the need to conduct a prospective randomized controlled trial to compare the efficacy of the two different doses on clinical and other virological end points in patients with HBV-related DC.