
Abstract
Microarray technology is highly used in biological research environments due to its ability to monitor the RNA concentration levels. The analysis of the data generated represents a computational challenge due to the characteristics of these data. Clustering techniques are widely applied to create groups of genes that exhibit a similar behavior. Biclustering relaxes the constraints for grouping, allowing genes to be evaluated only under a subset of the conditions. Triclustering appears for the analysis of longitudinal experiments in which the genes are evaluated under certain conditions at several time points. These triclusters provide hidden information in the form of behavior patterns from temporal experiments with microarrays relating subsets of genes, experimental conditions, and time points. We present an evaluation measure for triclusters called Multi Slope Measure, based on the similarity among the angles of the slopes formed by each profile formed by the genes, conditions, and times of the tricluster.
Introduction
Microarray technology is highly used in biological research environments due to its ability to monitor, for a great gene collection, the RNA concentration levels, thus enabling the study of genetic functions of species. 1 Bioinformatics and data mining have developed a vast number of computational tools that allow us to analyze data obtained using this technology and to find new knowledge that is hidden from human eyesight.2,3 One of the most studied approaches is pattern search in gene expression data. The genes exhibiting high correlation among their expression levels could be involved in similar regulatory processes. 4 The relationship between correlation and functionality has been proved in several studies as in the study by D'haeseleer et al. 5
Clustering techniques are suitable for performing pattern search by creating groups of genes that exhibit similar expression patterns. 6 Traditional clustering algorithms analyze the whole microarray dimensional space grouping genes taking into account all experimental conditions. 7 However, the activity of genes could only appear under a particular set of experimental conditions, exhibiting local patterns. Discovering these local patterns can be key to discover gene pathways, which could be hard to discover in other ways. For this reason, the paradigm of clustering techniques must be modified to methods that allow local pattern discovery in gene expression data. 8 Biclustering 9 addresses this problem by relaxing the conditions and by allowing assessment only under a subset of the conditions of the experiment, and it has proved to be successful in finding gene patterns.10,11
If a third dimension is added to the dataset besides genes and conditions, such as time, clustering and biclustering result insufficient. There is a lot of interest in temporal experiments because they allow an in-depth analysis of molecular processes in which the time evolution is important, for example, cell cycles, development at the molecular level, or evolution of diseases. 12 In this sense, triclustering appears as a technique going one step further by grouping genes under particular conditions and under particular time points, 13 thus being capable of managing three-dimensional (3D) data. Therefore, triclustering is suitable for the analysis of microarray experiments where several samples are taken at different time points. 14 This is of great interest since it allows for a deep analysis of biological processes where temporary development is important.
Both biclustering and triclustering attack NP-hard problems. 15 Therefore, algorithms based on heuristics are well suited to manage this kind of problem. In this sense, defining an appropriate quality measure for triclusters is an important and essential challenge. 16
In this work, we propose a quality measure called Multi Slope Measure (MSL), which measures the quality of a tricluster based on the similarity among the angles of the slopes formed by each profile formed by the genes, conditions, and times of the tricluster.
We show the results obtained applying the MSL measure embedded in the TriGen Algorithm, 17 an algorithm based on an evolutionary heuristic, genetic algorithms. The datasets used are a synthetic dataset and three real experiment datasets: the yeast cell cycle-regulated genes, 18 mouse degeneration of retinal cells, 14 and human transcription factor oncogene OTX2 silencing effect on D425 medulloblastoma cell line. 19
The results have been validated by three different methods. First, by analyzing the correlation among the genes, conditions, and times in each tricluster using two different correlation measures (Pearson 20 and Spearman 21 ). Second, by a graphic validation of the patterns extracted based on the graphic representation (see Graphic Representation subsection), and third we have provided functional annotations for the genes extracted from the Gene Ontology (GO) project. 22 The results obtained have been compared to two previously defined quality measures, MSR JD 23 and LSL, 24 showing improvement in the performance of the measure (see Results and Discussion section).
The rest of the article is structured as follows. A review of the latest related works can be found in State of the Art section. Methods section describes the MSL measure as well as a brief description of Triclustering, the graphic representation applied, and the TriGen algorithm. In the Results and Discussion section, we show the results and discussion of applying TriGen to the synthetic and real datasets. The last section shows the conclusions.
State of the Art
This section is to provide a general overview of recent works in the field of gene expression temporal data. In particular, for those works related to the application of triclustering, we focus on the measures applied to evaluate the triclusters.
We first present the authors’ previous contributions to this field. In our study, 23 we described MSR3D, an adaptation of the Mean Square Residue (MSR) 9 to the 3D space, so that a third factor, time in this case, can be taken into account. MSR3D measures the homogeneity of a tricluster in the relation of each value of the tricluster, with the average of all genes, average of all conditions, average of all times, average of all genes and conditions, average of all genes and times, average of all conditions and times, and average of all genes, conditions, and times in the tricluster. We also have presented LSL in our recent study, 24 which measures the quality of a tricluster based on the similarity among the slopes of the angles formed by the least square lines from each of the profiles formed by the genes, conditions, and times of the tricluster. LSL has obtained better results than MSR3D applied to the same datasets along with the TriGen algorithm. 24
Regarding other authors’ contributions, in 2005, Zhao and Zaki 25 introduced the triCluster algorithm to extract patterns in 3D gene expression data. They presented a measure to assess triclusters's quality based on the symmetry property. This allows for very efficient cluster mining since clusters are searched over the dimensions with the least cardinality.
g-triCluster, an extended and generalized version of Zhao and Zaki's proposal, was published one year later. 26 The authors claimed that the symmetry property is not suitable for all patterns present in biological data and proposed the Spearman rank correlation 21 as a more appropriate tricluster evaluation measure.
An evolutionary computation proposal was made by Liu et al. 27 The fitness function defined is a multiobjective measure that tries to optimize three conflicting objectives: clusters size, homogeneity, and gene-dimension variance of the 3D cluster.
LagMiner was introduced by Xu et al. 28 to find time-lagged 3D clusters, what allows in turn to find regulatory relationships among genes. It is based on a novel 3D cluster model called S 2 D 3 Cluster. They evaluated their triclusters on homogeneity, regulation, minimum gene number, sample subspace size, and time periods length.
Wang et al. 29 proposed a new algorithm called ts-cluster basing their definition for coherent triclusters also on finding regulatory relationships among genes. For that purpose, time shifting is also considered among time points in the evaluated triclusters.
A new strategy to mine 3D clusters in real-valued data was introduced by Sim et al. 30 The authors defined the Correlated 3D Subspace Clusters (CSCs), where the values in each cluster must have high co-occurrences and those co-occurrences are not by chance. They measure the clusters based on the correlation information measure, which takes into account both prerequisites.
Hu and Bhatnagar presented an approach focusing on the concept of Low-Variance 3-Cluster, 31 which obeys the constraint of a low-variance distribution of cell values.
The work by Liu et al. 32 was focused on finding Temporal Dependency Association Rules, which relate patterns of behavior among genes. The rules obtained are to represent regulated relations among genes.
Finally, a brief survey on triclustering applied to gene expression time series was published in 2011. 13 There are three main features that a triclustering algorithm can perform. According to Mahanta et al. 13 , these features are temporal coherence that makes reference to the ability of the algorithm to capture the coherence of different genes in a single time point across samples while generating the final triclusters and the ability to find triclusters with nonconsecutive time points and tricluster with a specific type of pattern (shifting, scaling, delayed). g-triCluster, 26 Moga3c, 27 LagMiner, 28 ts-cluster, 29 and Temporal Dependency Association Rules 32 perform the temporal coherence feature and only Tricluster 25 and Moga3c 27 perform finding triclusters containing non-consecutive time points. Tricluster 25 finds scaling patterns, LagMiner 28 finds shifting and scaling patterns and ts-cluster 29 focuses on time-delayed patterns; the rest do not focus on finding a specific type of pattern. Another feature examined is the algorithm type distinguishing between deterministic (Tricluster 25 , g-triCluster 26 ) and nondeterministic (Moga3c 27 ) approaches.
Methods
In this section, we describe our proposal, the tricluster quality measure called MSL that is based on tricluster's angular features. We will analyze all MSL principles and fundamentals and how it has been developed.
This section is structured as follows: Triclustering subsection describes the triclustering procedure as an evolution of its well-known predecessor biclustering. In the subsection Graphic Representation, we introduce the graphic representation, which is key for understanding the MSL measure. Then, in the subsection MSL Measure, we analyze the core of our work, the MSL measure. Finally, in the TriGen Algorith subsection, we briefly describe the TriGen algorithm.
Triclustering
Clustering techniques are applied to analyze gene expression data from microarray experiments. The dataset obtained from the experiment, D, contains genes and experimental conditions and clustering aims at finding subgroups of genes that share a behavior pattern according to their expression level. Biclustering appears as an evolution of clustering due to its ability to mine subgroups of genes and conditions from the data set D, where the genes exhibit highly correlated patterns of behavior under certain experimental conditions. 9
Triclustering emerges as an evolution of biclustering, taking into account the temporary evolution of genes under particular experimental conditions. In this way, from a dataset D obtained from a microarray experiment, which contains genes G D , conditions CD, and time points T D , we define triclustering as a technique that finds triclusters TRI1…,TRI n from D, where a tricluster TRI is formally defined as TRI = G × C × T, where G ⊆ G D , C ⊆ CD, and T ⊆ T D , 17 ie, a subset of genes that contains information related to the behavior of some genes from dataset G under conditions C at times T. Figure 1 shows a tricluster with genes as rows, conditions as columns, and time as depth.
Tricluster representation.
Graphic representation
In order to explain the MSL measure, we define the graphic representation of a tricluster TRI xop , with x, o, and p being either genes G, experimental conditions C, or time points T, so that the x elements in TRI xop will be on X axis and o elements in TRI xop will be the outlines represented in as many panels as p elements in TRI xop indicates, as can be seen in Figure 2.

Graphic representation of a tricluster.
To visually analyze the behavior patterns of a tricluster TRI, we always consider three graphical views:
TRI gct (x = G, o = C, p = T): one panel for each time, genes on the X axis, the expression levels on the Y axis, and the lines of conditions as the outline.
TRI gtc (x = G, o = T, p = C): one panel for each condition, genes on the X axis, the expression levels on the Y axis, and the time lines as the outline.
TRI tgc (x = T, o = G, p = C): one panel for each condition, times on the X axis, the expression levels on the Y axis, and the genes as the outline.
With TRI gct and TRI gtc , we can analyze how each gene expression level varies throughout conditions and times, respectively. TRI tgc represents how each gene varies throughout time for each condition.
MSL measure
After analyzing the graphic representation of a tricluster, we describe our proposal: the Multi Slope Measure (MSL). MSL measures the differences among the angles formed by every series traced on each of three graphic representations taking into account TRI gct , TRI gtc , and TRI tgc (subsection Graphic Representation). MSL takes into account the influence of neighboring time points. We can observe an example of TRI tgc view of TRI = G {g1, g4, g7, g10 C{c2, c5, c8} T{t0, t2 t11} in Figure 3. We can see how each outline or gene forms a set of angles (two for this particular example) defined by each time point in the X axis for every panel or experimental condition.

Angles for TRI tgc graphic view.
To calculate the MSL measure of a tricluster, we first perform the multiangular comparison term calculation. The multiangular comparison operation of a graphic representation xop from a tricluster TRI is defined in Equation 1a. We define AC
multi
of a tricluster's graphic representation TRI
xop
as the average of the differences δ of angles vectors







The AC
multi
. term is based on several operations with av
op
angle vectors. These elements have been obtained based on concept of series (Equation 2a) so that a series S
op
of a outline o for a panelp is a set of pair of values from the x axis (x
i
) and expression levels (el
j
) that form the outline. For each series S
op
, the alpha angle



To conclude, the MSL measure of a tricluster TRI (Equation 3) is the average of the angular comparison of the three graphic representations of the tricluster.
Following Figure 3, we show an example of AC
multi
(TRL
tgc
) calculation in Figure 4. First, we arrange the example tricluster TRI so that for each condition (panel) we obtain a table with one row per gene (outline) and one column per time point (X axis). Second, in order to get each av
gc
(Equation 1f) of angset (Equation 1b), we use Equation 2b from S
gc
series and obtain all angles
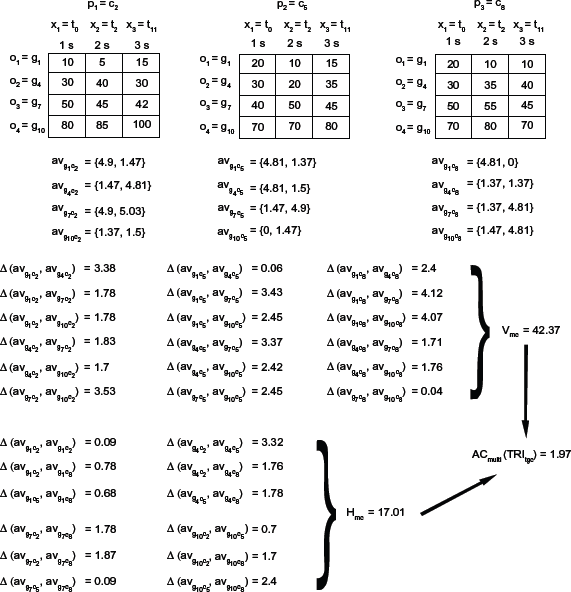
AC multi (TRI tgc ) example.

TriGen algorithm
In this section, we present the TriGen (Triclustering-Genetic based) algorithm, 17 where the MSL measure has been embedded in order to test its effectiveness. TriGen applies a bio-inspired paradigm of an evolutionary heuristic, genetic algorithms, in such a way that finds a set of triclusters from gene expression datasets where the time is also a component taken into account in the experiment. This method mimics the process of natural selection by creating an initial population of individuals representing solutions that are crossed and mutated for a number of generations, with the best individuals in the population being finally selected. The MSL measure has been applied as the fitness function to assess the quality of the triclusters or solutions in the population. The flowchart of the TriGen algorithm can be seen in Figure 5. We now define the most important elements of the algorithm such as inputs, outputs, codification of individuals, and genetic operators.

TriGen algorithm flowchart.
Input: The TriGen algorithm has two input arguments:
D: A dataset containing the gene expression values from a microarray experiment containing genes D G , experimental conditions D C , and times D T . Therefore, each cell [i,j,k] from D where i ∊ D G , j ∊ D C , and k ∊ D T , represents the expression level of the gene i under the experimental condition j at time k.
P: Set of parameters to execute the algorithm as described in Table 1. These parameters control the number of solutions or triclusters to find (N), the number of generations to execute (G), the number of individuals in the population (I), and the randomness factor they are generated with the initial population (Ale) as well as weights for the selection and mutation operators (Sel and Mut), weights to control the effect of the MSL measure (w f ), the size of the triclusters (w g , w c , w t ), and weights to control the overlap among solutions (w g , w c , w t ).
TriGen algorithm parameters.

The parameters have been chosen after an exhaustive experimentation with all possible ranks of values. Each of the parameters has effect on the triclusters found: size, overlapping, exploration versus exploitation of the algorithm, etc. We now describe the effects of each of the parameters. In the execution of TriGen, each parameter is associated to a genetic operator, that is, G controls the whole evolutionary process so an increase in the number of generations implies a greater number of recombination of individuals. Therefore, an excessive increase in G may favor exploitation versus exploration in excess and the algorithm may return solutions that fall into a local minimum. I and Ale control the initial population creation, and when the number of individuals I is increased, a larger search space for the solutions is created so that an excessive increase can create a scatter search effect, and therefore, not return good quality solutions; an increase of the randomness rate Ale in the initial population has to be combined with the overlap control to make sure that a wide area of the space of solutions is initially covered. Sel controls the selection mechanism, and as a result, the crossover and a high Sel creates individuals with a low level of genetic recombination, favoring exploitation versus exploration, and if the parameter is increased in excess, the algorithm may fall into a local minimum. On the contrary, a high probability of mutation Mut favors exploration versus exploitation, and if increased in excess, we will end up with solutions in many areas of the search space but with low quality levels. w and wo combined with w f control the fitness function; w weights control the number of items in the solutions, an increase of these weights involves favoring solutions with more volume; the increase of wo weights leads to little or nonoverlapped solutions, an excessive increase can lead to the loss of interesting solutions. The content under Genetic Operators in the subsection TriGen Algorithm explains all operators and how a parameter variation affects the execution of TriGen.
Output: The TriGen algorithm's output will be a set of N triclusters, formally SOL = {TRI1, TRI2,…, TRI N }. Each TRI i ε SOL is composed of a subset of genes TRI G , conditions TRIC, and times TRI T from the input dataset D and has the best score in its population when evaluated under the MSL measure.
Codification of individuals: Each individual in the evolutionary process of the TriGen algorithm represents a tricluster, which is a potential solution. Therefore, an individual is represented as a subset of genes
Overlapping control: We have designed an overlapping control mechanism to avoid overlapping among the tri-cluster solutions obtained. It is called Data Hierarchy and consists in maintaining the number of occurrences of genes, conditions, and time points of dataset D in each tricluster solution from less to most visited in such a way that, as we will explain in Initial Population in the subsection TriGen Algorithm, the initial population creation uses this structure to initialize population with the minimum overlapping. This Data Hierarchy is updated at every generation a new tricluster solution is selected.
Genetic operators:
Initial population: With the initial population method, I individuals are generated attending to the Ale randomness parameter. An Ale percent of individuals are created at random by two methods: half of the individuals are purely randomly generated, this is, a random subset of genes TRI G , conditions TRI C , and times TRI T are chosen from D and the other half is also randomly created but controlling that the values for the genes TRI G are contiguous, the values for the conditions TRI C are contiguous and the times TRI T are contiguous as well. The rest of the individuals are randomly created, but taking into account the previously created individuals to control overlapping of solutions according to Data Hierarchy structure (Overlapping Control in the subsection TriGen Algorithm).
Fitness function: Our proposed measure has been included as the genetic algorithm's fitness function FF(TRI) along with the size and the overlapping control. As can be seen in Equation 4, MSL has been combined with six other factors as a weighted average. Three of these factors 
Selection: Three groups of individuals are randomly selected sorted from lowest to highest according to the fitness function, and then a random selection from the three groups is made. The Sel parameter indicates how many of these individuals will pass to the next generation. The rest of the individuals until completing the next population (I – #Setected individuals) will be created based on the crossover operator.
Crossover: To complete the next generation, we create new individuals with this operator as follows: two individuals (parents, A and B) are combined to create two new individuals (offsprings, chitdl and chitd2). The parents are randomly chosen. Their genetic materials are combined by a random one-point cross in the genes TRI G , conditions TRI C , and time TRI T and mixing the coordinates in both children. 17
Mutation: An individual can be mutated according to a probability of mutation, Mut. The mutation probability is verified for every individual, and if it is satisfactory, one out of nine possible actions is taken. These actions are add a new random gene to TRI G , add a new condition to TRI C , or add a new time point to TRI T , by removing a random gene, condition, or time, or by changing a random gene or condition for another that is randomly chosen. The election of these actions is also random. For the case of addition of a new gene, condition, or time, the operator checks whether the new member is already in the individual or not.
Results and Discussion
In this section, we show the results obtained by application of MSL as a fitness function embedded in the TriGen algorithm 17 (see the subsection TriGen Algorithm).
MSL has been applied to four different datasets: one synthetically generated dataset and three real datasets. The real datasets are obtained from experiments with the yeast cell cycle (Saccharomyces cerevisiae), 18 an experiment with mice (Mus muscutus) called GDS4510 14 and data from experiments with humans (Homo sapiens) called GDS4472. 19 The last two data sets have been retrieved from Gene Expression Omnibus, 33 a database repository of high-throughput gene expression data. All biological experiments examine the behavior of genes under conditions at certain times.
For the analysis of the triclusters obtained as the result of the experiments, we have developed a three-step process based on providing information related to correlation among expression values, the graphic properties of the representation of the values, and biological validation.
The correlation validation is based on the Pearson and Filon
20
and Spearman
21
coefficients. For every tricluster, we calculate the average of the correlation coefficients between each combination of gene, condition, and time for all genes. For instance, for a tricluster with four genes {1, 4, 8, 10}, two conditions {3 and 7}, and three times {1, 3 and 5}, we provide the Pearsons and Spearmans correlation coefficient average for values at the eight possible combinations, each having three time points:
The graphic properties of the representation are shown as described in the subsection Graphic Representation as a way to visually check how the gene patterns behave.
Finally, for the biological validation, we will show the GO terms 22 related to the triclusters. We present a GO analysis table in which we include the most representative terms extracted by the Ontologizer software, 34 each term associated to a P-value that denotes the relevance level of the term. In this type of studies, P-values are considered as relevant below 0.05 and are better when closer to 0. Regarding the GO project, it is a major bioinformatics initiative with the aim of standardizing the representation of gene and gene product attributes across species and databases. The project provides an ontology of terms for describing gene product characteristics and gene product annotation data. The ontology covers three domains: cellular component, the parts of a cell or its extracellular environment; molecular function, the elemental activities of a gene product at the molecular level, such as binding or catalysis; and biological process, operations, or sets of molecular events with a defined beginning and end, pertinent to the functioning of integrated living units: cells, tissues, organs, and organisms.
We have compared the results obtained to those from Gutiérrez-Avilés and Rubio-Escudero, 23 where the fitness function was the MSR 3D measure and also to the results in Gutiérrez-Avilés and Rubio-Escudero, 24 where the fitness function was the LSL measure. The comparison has been made in terms of correlation and GO analysis. For each real experiment, we have compared the maximum, minimum, and mean Pearson's and Spearman's correlation index and the maximum, minimum, and mean P-value for each solution considered.
All experiments were executed on a multiprocessor machine with 64 processors, Intel Xeon E7-4820 2.00 GHz with 8-GB RAM memory. We have used Java to implement TriGen algorithm (and other ad hoc developments) and an R framework to create graphics and get dataset resources from GEO. 33
We now analyze the results obtained in each of the four experiments.
Synthetic experiments
Synthetic data are widely used not only for testing the performance of microarray analyzing techniques 14 but also in more general data mining publications. 35 It has the advantage that the process that generated the data is well known and so one is able to judge the success or failure of the algorithm. 36
In this work, we have used an application designed by ourselves to generate the synthetic data used in this experiment. We have executed the TriGen algorithm with the MSL measure over a synthetic dataset composed of 4,000 genes, 30 experimental conditions, and 20 time points whose expression levels were randomly generated by a cryptographic secure standard library Math3 provided by Apache Commons. 37 In this dataset, we inserted 10 triclusters composed of 150 genes, 6 experimental conditions, and 4 time points, whose expression levels form a constant behavior pattern. These triclusters are located in random positions in the dataset.
To see the behavior of the MSL measure applied along with TriGen and also with the aim of analyzing the effect of the value of the parameters in the solutions, we have made executions setting N to 200 and varying other control parameters as follows: G ε {100, 200}, I ε {50, 100}, Sel ε{0.5}, Mut ε{0.2, 0.3}, Ale ε {0.3, 0.5}, w g ε {0.03, 0.05, 0}, w c ε{0, 0.01}, w t ε{0,0.01}, wo g ε{0.04, 0.05}, wo c ε {0,0.03}, and wo t ε {0,0.03} (see Input in the TriGen Algorithm subsection for a detailed description of these parameters). The algorithm has been capable of finding between 94% and 100% of the inserted triclusters. There were no false positives or false negatives found. The application of the MSR 3D along with the TriGen algorithm in the study by Gutiérrez-Avilés and Rubio-Escudero 23 was capable of finding 91% to 95% and LSL 24 obtained a matching ratio between 93% and 97% of the triclusters, so we can see in slight improvement when applying the MSL measure.
Yeast elutriation experiments
For this experiment, we have applied the TriGen algorithm with MSL measure to the yeast (Saccharomyces cerevisiae) cell cycle problem, 18 specifically the Elutriation experiment. The yeast cell cycle analysis project's goal is to identify all genes whose mRNA levels are regulated by the cell cycle. The resources used are public and available in http://genome-www.stanford.edu/cellcycle/. Data have been normalized as part of the preprocessing. We have created a dataset Delu 3D from the elutriation experiment with 7,744 genes, 13 experimental conditions, and 14 time points. Experimental conditions correspond to different statistical measures of the Cy3 and Cy5 channels while time points represent different moments of taking measures from 0 to 390 minutes.
The parameter configuration used for this experiment is shown in Table 2. We set G and I values in order to obtain a default exploration of the solution space and Ale, Sel, and Mut provide us with a high random factor in population generation, low elitism in the next generation promotion, and high mutation factor, respectively. We favor solutions with a high number of genes setting w g to 0.05 and solutions with high variability in genes, conditions, and times thanks to wo g , wo c , and wo t , set to 0.05. These configurations have been obtained as a result of a deep experimental study on the Delu 3D dataset.
TriGen algorithm control parameters for yeast cell cycle dataset.

Regarding the correlation analysis, we can observe in Table 3 show Pearson and Spearman's values vary between [0.95,0.97] and [0.98,1], respectively, which implies a high correlation between genes series for every experimental condition through time points. These high values confirm us that the quality of the triclusters obtained from these experiments is very high in terms of correlation.
correlation results for triclusters from the yeast cell cycle dataset.

We can see the graphic representation of tricluster TRI11 in Figure 6. Only one out of the 20 triclusters obtained has been represented for legibility reasons. We can observe how TRI11 with 200 genes, 2 conditions, and 3 time points shows a coherent pattern through time points for CH1I and CH1B experimental conditions (TRI gct view, Fig. 6C). TRI gct (Fig. 6A) and TRI gtc (Fig. 6B) show how conditions vary for 0 minutes, 330 minutes, and 390 minutes for all genes and how times vary for CH1I and CH1B for all genes, respectively. In Figure 6A we can see that each of the two conditions vary in almost the same way for every time point. This can also be observed in Figure 6B but according to time variation for every experimental condition.

TRI11 graphic representations from yeast cell cycle results. (A) Sample curves, (B) time curves, (C) gene curves.
Finally, regarding the experiment biological analysis, we can see GO results of the biological annotations related to the genes selected in TRI11 (Table 4). We can see how three related biological processes have been annotated with TRI11 genes, these are cellular amide catabolic process (GO:0043605), allantoin metabolic process (GO:0000255), and catabolic (GO:0000256) process. These biological processes are related to a more general one called cellular amide metabolic process (GO:0043603) in which individual cells carry out the chemical reactions and pathways involving an amide. We can observe how two molecular functions as RNA-DNA hybrid ribonuclease activity (GO:0004523) and endonuclease activity with either ribo- or deoxyribonucleic (GO:0016893) have been annotated for TRI11 as well. Both groups of terms, biological processes, and molecular functions are related with the metabolic process of the cell. All terms annotated have a high level of statistical significance denoted by P-values in the [1.98E-09,1.01E-03] interval.
GO analysis for tricluster TRI11 found in the yeast cell cycle dataset.

We can observe in Table 5 a comparison among MSR3D, LSL, and MSL measures according to the published Elutriation results published in the studies by Gutiérrez-Avilés and Rubio-Escudero23,24 Regarding correlation indexes, a clear improvement of MSL in relation to MSR3D and LSL can be observed as well as an improvement in terms of the P-value.
Comparison of MSR3D, LSL, and MSL yeast cell cycle results.

Mouse GDS4510 experiments
In this experiment, we have used a dataset obtained from the GEO 33 with accession code GDS4510 whose title is rd1 model of retinal degeneration: time course. 14 In this biological experiment, the degeneration of retinal cells in different individuals of home mice (Mus musculus) is analyzed over 4 days just after birth, specifically on days 2, 4, 6, and 8. Data have been normalized as part of the preprocessing. We have formed our input dataset DGDS4510 3D with 22,690 genes, 8 experimental conditions (one for each individual involved in the biological experiment), and 4 time points. Each of the replicates has been treated as an independent condition.
The parameter configuration used for this experiment is shown in Table 6. We set G to a default value and I is increased in order to expand the exploration of the solution space, Ale, Sel, and Mut have been set to 0.5 in order to get a medium rate of randomness factor, elitism, and mutation rates, respectively. With this size and overlapping control, we use TriGen to find solutions with a high number and variability of time points. This configuration has been obtained as a result of an extensive experimental study over DGDS4510 3D dataset.
TriGen algorithm control parameters for mouse GDS4510 dataset.

According to Table 7, we can see how Pearson and Spearman's values vary between [0.54,0.96] and [0.56,0.9], respectively. Pearsons values fall in the [0.8,0.9] interval are predominant in most of the solutions as well as Spearman's ones. This fact implies a high correlation between gene series for every experimental condition through time points in most of the solutions of this experiment.
Correlation results for tricluster mouse GDS4510 dataset.

In Figure 7 we show three graphic representations of tri-cluster TRI10 composed by 50 genes, 2 experimental conditions, and 4 time points. In the TRI tgc graphic representation, we can see how all genes form an almost perfectly coherent pattern through the four time points for all experimental conditions. This fact is reinforced when we observe TRI gct (Fig. 7A) and TRI gtc (Fig. 7B) graphic representations in which variation of expression levels in sample and time perspectives show coherent patterns.

TRI10 graphic representations from mouse GDS4510 results. (A) Sample curves, (B) time curves, (C) gene curves.
We can see the biological validity of TRI10 in Table 8. It reflects very good results with P-values in the [7.92E-30,7.52E-07] interval. Terms like olfactory receptor activity (GO:0004984), which is the molecular function that denotes the transmission of the signal from both sides of cellular membrane in order to initiate a change in cell activity due to detection of smell, has been annotated close to sensory perception of chemical stimulus (GO:0007606) and detection of chemical stimulus involved in sensory perception of smell (GO:0050911), which have a very low P-value, are biological processes that have olfactory receptor activity as a part of them. We can see how pheromone receptor activity (GO:0016503), that involves the effects of pheromone in cell activity, is related with G-protein-coupled receptor signaling pathway (GO:0007186) since it is a biological process whose pheromone receptor activity is part of this aforementioned term through G-protein coupled receptor activity (GO:0004930) term that has been annotated as well.
GO analysis for tricluster TRI10 found in the mouse. GDS4510 dataset.

Regarding the comparison with MSR 3D and LSL measures (see Table 9), we can see that MSL performs slightly better than MSR 3D and very similar to LSL, which performs exceptionally well for this particular experiment, in terms of correlation indexes. In terms of GO analysis, we can see a marked improvement of MSL against MSR 3D and the minimum P-value of the LSL measures.
Comparison of MSR 3D , LSL and MSL GDS4510 results.

Human GDS4472 experiments
For this experiment, the DGSD4472 3D dataset has been built from source data obtained from GEO 33 under code GDS4472 titled Transcription factor oncogene OTX2 silencing effect on D425 meduttobtastoma cell line: time course. 19 In this experiment, the effect of doxycycline on medulloblastoma cancerous cells at six time points after induction for 0, 8, 16, 24, 48, and 96 hours has been analyzed. Data have been normalized as part of the preprocessing. DGSD4472 3D is composed of 54,675 genes, 4 conditions (one for each individual involved), and 6 time points (one per hour).
We can see the parameter configuration for TriGen used for this experiment in Table 10. We want to expand the search space setting G to 700 and I to 500 due to the size of the input dataset, we increase the random factor setting Ale to 0.8 and we combine low elitism with high-variability setting Sel to 0.2 and Mut to 0.9, respectively. Regarding size control, we can see how we want triclusters with a balance between a high number of genes, w g to 0.01, and a high number of conditions and times, w c to 0.045 and w t to 0.045, respectively. The overlapping control follows the same proportion with wo g set to 0.01, wo c set to 0.045, and wo t set to 0.045 in terms of variability of genes, experimental conditions, and time points of the solutions.
TriGen algorithm control parameters for human GDS4472 dataset.

As regards the correlation analysis, we can observe in Table 11 how Pearson and Spearman's values vary between [0.47,0.96] and [0.45,1], respectively, and most of the solutions found have both index above 0.9; therefore, this is a good experiment in terms of correlation.
Correlation results for tricluster human GDS4472 dataset.

According to graphic representation, we show the tri-cluster TRI19 in Figure 8, which has 40 genes, 4 experimental conditions, and 5 time points. We can see a coherent behavior pattern for all genes through five time points for every experimental condition in the TRI tgc graphic representation (Fig. 8C). In the TRI gct graphic representation (Fig. 8A), we can see a homogeneity variation of expression levels of all genes for all experimental conditions at each time point. We can also see this fact in TRI gtc graphic representation (Fig. 8B).

TRI19 graphic representations from human GDS4472 results. (A) Sample curves, (B) time curves, (C) gene curves.
We can see the biological analysis of TRI19 in Table 12 in which there is a set of terms annotated with P-value in [3.33E-60,5.99E-48] interval that is a very low rate of P-value; therefore, they are good biological results. We pay attention to term GO:0006614 named SRP-dependent cotranslational protein targeting to membrane that describes the targeting of proteins to a membrane that occurs during translation and it is related to another two biological processes annotated as well: cotranslational protein targeting to membrane (GO:0006613) and protein targeting to ER (GO:0045047). We also underline the GO:0019083 term named viral transcription that describes the process by which a viral genome is transcribed within the host cell that is closely related with viral gene expression (GO:0019080) biological process since the transcription process is part of the gene expression one. Finally, we can see how nuclear-transcribed mRNA catabolic process, nonsense-mediated decay, GO:0000184 term, is annotated beside nuclear-transcribed mRNA catabolic process (GO:0000956) and mRNA catabolic process (GO:0006402) whose biological processes define the first one.
GO analysis for tricluster TRI19 found in the human GDS4472 dataset.

Regarding to the comparison between MSL and the other two measures, MSR 3D and LSL (Table 13) we can see an improvement of MSL from MSR 3D and LSL in terms of maximum correlation indexes, this fact is less perceptible in terms of minimum and mean correlation indexes. We can also appreciate a high improvement of MSL over MSR3D and LSL in terms of three considered aspects of P-value. Therefore, we can affirm that MSL globally improves the other two measures.
GO analysis for tricluster TRI19 found in the human GDS4472 dataset.

Conclusion
In this work, we have presented a new evaluation measure for triclusters, MSL, which measures the homogeneity among genes, conditions, and times in a tricluster. We have analyzed how this measure has been formulated in detail and we have also applied it as a part of TriGen algorithm, 17 which is our triclustering approach to mine triclusters from microarray experiments involving time, in order to assess the quality of the measure.
We have applied MSL embedded in the TriGen algorithm in four datasets: synthetically generated data, data from experiments with the yeast cell cycle (Saccharomyces cerevisiae) obtained from the Stanford University
18
and two datasets retrieved from Gene Expression Omnibus,
33
an experiment with mice (Mus muscutus) and an experiment with humans (Homo sapiens). All experiments examine the behavior of genes under conditions at certain times. The results obtained in real experiments have been validated by a three-way analysis: first analyzing the correlation among the genes, conditions, and times in each tricluster using two different correlation measures: Pearson
20
and Spearman,
21
second graphical analysis and finally providing functional annotations for the genes extracted from the GO project.
22
We have provided a comparison among MSL and another two developed measures
Summarizing, regarding synthetic experimental results, we can observe how MSL combined with TriGen has been capable to extract from 94% to 100% of the triclusters. Regarding the real dataset results, they are also successful, with correlation values close to 1, good graphical representations in which you can distinguish very clear behavior pattern and GO validation with high levels of significance for the terms extracted (P-values smaller than 0.05 and very specific terms).
MSL is a tricluster evaluation measure created to assess the quality of triclusters extracted from temporal experiments with microarrays, but it can be used in other biologically related fields, for instance combining expression data with gene regulation information by means of substituting the time dimension by ChIP-chip data representing transcription factor-gene interactions what can provide us with regulatory network information. This proposal can also be applied to mine RNA-seq data repositories. Triclustering can also be applied to not biologically related fields, for instance, the seismic zonification of areas at risk of undergoing an earthquake. 38 In this case, the third component does not identify time points but features associated to every pair of geographical coordinates of the area under study.
The next step in our researching work is to gather all aspects of tricluster experimental evaluation presented in Results and Discussion section, that is correlation, graphic representation, and biological validation, in one measure, and thus, improving the experimental workflow of triclustering.
Author Contributions
Conceived and designed the experiments: DG-A, CR-E. Analyzed the data: DG-A. Wrote the first draft of the manuscript: DG-A. Contributed to the writing of the manuscript: DG-A, CR-E. Agree with manuscript results and conclusions: DG-A, CR-E. Jointly developed the structure and arguments for the paper: DG-A, CR-E. Both authors reviewed and approved of the final manuscript.