
Abstract
To better understand the transmission and evolution of Mycobacterium tuberculosis (MTB) in Taiwan, six different MTB isolates (representatives of the Beijing ancient sublineage, Beijing modern sublineage, Haarlem, East-African Indian, T1, and Latin-American Mediterranean (LAM)) were characterized and their genomes were sequenced. Discriminating among large sequence polymorphisms (LSPs) that occur once versus those that occur repeatedly in a genomic region may help to elucidate the biological roles of LSPs and to identify the useful phylogenetic relationships. In contrast to our previous LSP-based phylogeny, the sequencing data allowed us to determine actual genetic distances and to define precisely the phylogenetic relationships between the main lineages of the MTB complex. Comparative genomics analyses revealed more nonsynonymous substitutions than synonymous changes in the coding sequences. Furthermore, MTB isolate M7, a LAM-3 clinical strain isolated from a patient of Taiwanese aboriginal origin, is closely related to F11 (LAM), an epidemic tuberculosis strain isolated in the Western Cape of South Africa. The PE/PPE protein family showed a higher dn/ds ratio compared to that for all protein-coding genes. Finally, we found Haarlem-3 and LAM-3 isolates to be circulating in the aboriginal community in Taiwan, suggesting that they may have originated with post-Columbus Europeans. Taken together, our results revealed an interesting association with historical migrations of different ethnic populations, thus providing a good model to explore the global evolution and spread of MTB.
Introduction
Mycobacterium tuberculosis (MTB) continues to be a leading cause of human deaths by an infectious agent. It has been estimated that approximately one-third of the world's population has been infected with the tubercle bacillus, and 1.5 million die from MTB infection every year. 1 In 2012, 12,338 new cases were reported in Taiwan, with an estimated annual incidence of 53 cases per hundred thousand people. 2 Epidemiologic studies have revealed that different genotypes of MTB may be prevalent in different geographic regions worldwide and that genotype distribution is closely associated with population migrations.3–5 The distribution of human MTB genotypes is closely associated with geography, ethnicity, age, and host factors. Recent developments in DNA sequencing technologies have revolutionized tuberculosis (TB) research, contributing to major advances in understanding the evolution and pathogenesis of MTB and facilitating the development of new diagnostic tests with increased specificity for TB. Identification of the genomic features of major MTB strains is key to deciphering the transmission of virulence and drug resistance among different strains. Thus, comparative analysis of whole-genome sequences can provide better insights into the evolution of the MTB strains present in Taiwan. Achieving these goals will improve our understanding of the epidemiology of TB in Taiwan and help guide prevention policy.
Taiwan, a relatively isolated island in the southeast of mainland China, is regarded as a mixing vessel of immigrants over the past four centuries as colonization by different waves of ethnic groups occurred. The aborigines in Taiwan are of Austronesian descent, which distinguishes them from the major ethnic group on the island, the Han Chinese, and now reside predominantly in the mountainous regions or rural areas. 6 Documentation of aboriginals on the island can be traced back to the 16th century, when Spanish sailors arrived and named the island. There are 12 aboriginal tribes on the island, which are presumed to represent the ancestral colonies that inhabited the island for at least 4000 years. The other two predominant ethnic populations of Taiwan are descended from Han Chinese who migrated to the island in two major waves: the first during the Ming Dynasty around 1600 and the second between 1945 and 1950, when members of the military, veterans, and some civilians emigrated from mainland China due to the civil war there 7 ; in total, about two million mainland Chinese have migrated to Taiwan to date. Taiwan was occupied by the Dutch for 40 years beginning 1660, and the Japanese from 1895 until 1945.
We previously demonstrated that the Beijing ancient strain and the Haarlem strain are the predominant MTB strains infecting aborigines in eastern Taiwan (Hualien City), the East-African Indian (EAI) strain is prevalent in southern Taiwan aborigines, and the Beijing modern strain is predominant in Han Chinese.7–10 In the present study, six MTB strains – isolates of the Beijing ancient sublineage, the Beijing modern sublineage, Haarlem, EAI, T1, and Latin-American Mediterranean (LAM) – representing the major types of clinical strains isolated from three different ethnic groups (aboriginals, Han Chinese, “veterans”) in Taiwan7,8 were subjected to whole-genome sequencing.11–13 The six Taiwan genomes were then compared to four reference MTB strains (H37Rv, H37Ra, CDC 1551 (LAM), and F11 (LAM)14–16) as well as the genome of Mycobacterium bovis. The presence of significant sequence diversity in MTB could provide a basis for understanding pathogenesis, immune mechanisms, and bacterial evolution. Polymorphic genes are good candidates for virulence and immune determinants, because proteins that interact directly with the host are known to have elevated divergence. Examples in MTB are the PE/PPE genes that encode proteins with proline-glutamate and proline–proline–glutamate motifs. 17
Methods
Study patients and bacterial isolates
Aborigines and veterans are entitled to government-subsidized health benefits under the National Health Insurance Claim System of Taiwan; therefore, the identities of these patients were confirmed by the type of insurance policy or the type of identification card. We obtained MTB isolates from three hospitals in three different regions of Taiwan. Patients with symptoms compatible with pulmonary TB and with sputum cultures positive for M. tuberculosis complex were included. Isolates were stored frozen by the participating hospitals and sent to the TB laboratory in the Division of Infectious Diseases of National Health Research Institutes (NHRI). MTB genomic DNA was extracted from primary LJ egg cultures as described previously. 18 In this study, we first characterized the phenotypes and genotypes of at least 1000 isolates of MTB from different ethnic populations, at least 100 isolates from each population (aborigine, veterans, and Han Chinese), followed by a comparative genomics study to provide a snapshot of mycobacterial evolution and its pathogenesis.
Genotyping
All isolates were subjected to spoligotyping (ST) and determination of the 19 variable number of tandem repeats (VNTR)-mycobacterial interspersed repetitive unit (MIRU) loci. ST was performed as previously described by Kamerbeek et al. 19 Data were compared with the international SpolDB4.0 database. 20 VNTR-MIRU loci (ETR-A, B, C, D, E, F; MPTR-A; MIRU-2, 4, 10, 16, 20, 23, 24, 26, 27, 31, 39, 40) were individually amplified and analyzed as previously described by Supply et al. 21 Results from each of the 19 loci were combined to create 19-digit allelic profiles.
Genome sequencing and DNA analysis
MTB strains were sequenced separately to 10- to 30-fold coverage of the genome using a Genome Sequencer 20 (GS20) or a Genome Sequencer FLX (GS FLX) instrument (454 Life Sciences, Roche) 22 with a 500-800-base pair shotgun library for each strain. The reads generated by 454 pyrosequencing were assembled using the GS De Novo Assembler version 2.5.3 provided by the manufacturer. Protein-coding genes were predicted from all contigs of each assembly using GLIMMER version 3.02. 23 All contig sequences of the six strains were compared to the reference strain H37Rv to detect single-nucleotide polymorphisms (SNPs) using MUMmer version 3.20. 24 The genome sequence of H37Rv was downloaded from the NCBI ftp (ftp://ftp.ncbi.nih.gov/genomes/Bacteria/Mycobacterium_tuberculosis_H37Rv_uid57777/). MUMmer provides a pipeline with the programs “nucmer”, “delta-filter”, and “show-snps” for sequence aligning, repeat filtering, and SNP/INDEL calling. The pipeline is able to detect all SNPs between two genomes, even where there are many sequence rearrangements. Phylogenies based on the whole genomes of the six MTB strains sequenced in this study and the five completely sequenced strains available through NCBI were constructed using the MUMmer distance matrix method adopted by Chan et al. 25 In brief, MUMs (maximal unique matches between the two genomes) obtained from MUMmer were summarized for each pair of genome sequences and then divided by the smaller genome length of the pair.
Construction of phylogenetic trees
The genetic diversity (based on MIRU-VNTR loci and ST) of the six selected Taiwan MTB strains was used to build a neighbor-joining tree. A pairwise distance can be obtained after log transformation and multiplication by -1. After the distance matrix of all the 11strains (the six Taiwan isolates and five reference strains) was completed, the program MEGA4.1 was applied to build a phylogenic tree using the minimum-evolution method (Fig. 1C).
Genetic diversity of six Taiwan MTB strains. Neighbor-joining tree showing genetic diversity of six Taiwan MTB strains based on MIRU-VNTR loci (A) and spoligotyping (B). A whole-genome sequencing-based dendrogram was generated using minimum evolution methods (C).
Screening of SNPs in the MTB genome
SNP identification in strains A18, A27, M3, M7, M24, and W6 using H37Rv as the reference.

Statistical analyses
BioNumerics software (v 3.0; Applied Maths) was used to analyze MIRU by character types. Similarities between MIRU types were calculated using the categorical coefficient, in which all MIRU loci were weighted equally. This procedure counted the number of matched loci between pairs of isolates; when there was a difference, they were scored as unmatched, irrespective of the number of repeats present (thus, 1 versus 3 scored the same, ie, unmatched, as 1 versus 4). A dendrogram was constructed by the unweighted pair group method using arithmetic means averages. MIRU types were discriminated by similarity index and compared with octal format, ST, and family results.
Ethics statement
This study was approved by the Human Ethics Committee of the National Health Research Institutes, Taiwan (Code: EC0961103). Because of the retrospective nature, routine collection of clinical data in daily practice, and dislinkage of personal information, the requirement to obtain informed consent was waived by our institutional review board.
Results
Sequencing of six M. tuberculosis genomes from patients in Taiwan
Characteristics of the six Taiwan strains sequenced in this study.

Genome assemblies of six Taiwan M. tuberculosis strains.
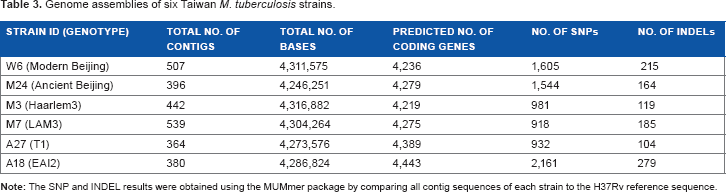
Pairwise comparison of the M. tuberculosis genomes
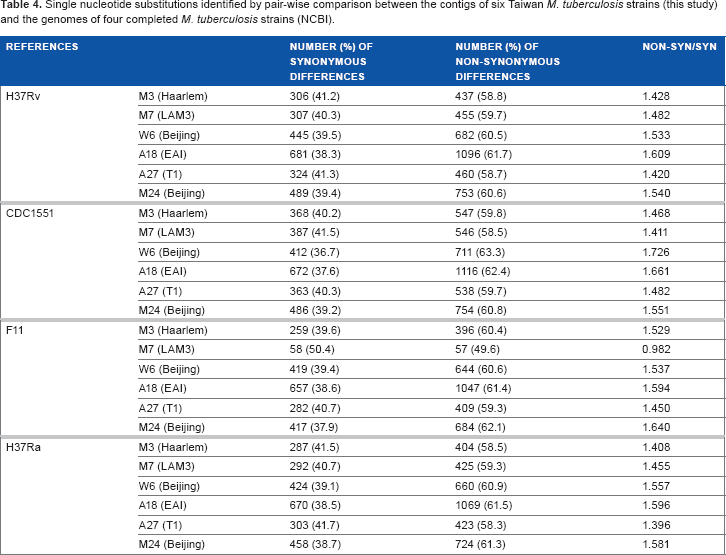
Single nucleotide substitutions identified by pair-wise comparison between the contigs of six Taiwan M. tuberculosis strains (this study) and the genomes of four completed M. tuberculosis strains (NCBI).
Phylogeny of MTB strains based on genomic sequencing and VNTR-based genotyping
The six Taiwan MTB strains and the five reference strains were included in this analysis (Fig. 1). This phylogenetic tree (Fig. 1B) is almost congruent with the one we constructed using the neighbor-joining method as well as with the 17 loci MIRU-based analysis (Fig. 1A).
SNP identification in strains A18, A27, M3, M7, M24, and W6 using H37Rv as the reference
The six strains have similar N/S ratios when all SNPs in the protein-coding genes are taken into consideration. It is notable that three of the strains (A27, M7, and M3) have fewer SNPs and may be evolutionarily more closely related to H37Rv. The genes for the PE/PPE family proteins in A27, M3, and M7 also have fewer SNPs in comparison to the other strains. The dn/ds ratio for the PE/PPE family was higher in A18 (EAI2_Manilla strain) compared to the average dn/ds ratio for the PE/PPE family.
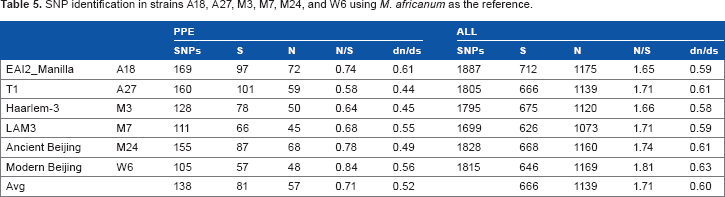
SNP identification in strains A18, A27, M3, M7, M24, and W6 using Mycobacterium africanum as the reference
SNP identification in strains A18, A27, M3, M7, M24, and W6 using M. africanum as the reference.
SNP identification in strains A18, A27, M3, M7, M24, and W6 using Mycobacterium cannettii and Mycobacterium marinum as references
SNP identification in strains A18, A27, M3, M7, M24, and W6 using M. cannetti as the reference.

SNP identification in strain A18, A27, M3, M7, M24, and W6 using M. marinum as the reference.

Analysis of M. bovis genomic SNPs using M. marinum as the reference
We extracted the sequence data for the 3953 coding sequences from the M. bovis genome (accession no. BX248333) and mapped them to the M. marinum genome. In total, 231,941 SNPs were identified in protein-coding genes and, among these, 49,900 are nonsynonymous substitutions. The N/S ratio was determined to be 0.27 for all protein-coding genes. A total of 1104 of the SNPs belong to PE/PPE family protein genes, of which 315 are nonsynonymous substitutions. The N/S ratio for PE/PPE family protein genes was determined to be 0.399. For the PE/PPE family or all genes, the N/S ratio for the SNPs identified in M. bovis using M. marinum as the reference is <1. This is very different from the previous analysis for the six Taiwan MTB strains against M. marinum, in which the SNPs of the PE/PPE family have a higher N/S ratio than that of all genes (average 0.58 for PE/PPE versus 0.27 for all).
Discussion
Comparison of nonsynonymous substitutions per nonsynonymous site to the number of synonymous substitutions per synonymous site (dn/ds) between homologous genes is an important index in molecular evolution. In this study, we compared the dn/ds ratios among the SNPs called from mapping the sequencing reads of six MTB strains to different reference genomes. 17 There are two types of natural selection in biological evolution: (1) positive selection promotes the spread of beneficial alleles and (2) negative selection hinders the spread of deleterious alleles. 27 In our results, the average dn/ds values for SNPs called using H37Rv or M. africanum were found to be much higher than those using M. cannettii and M. marinum as references. Apparently, higher dn/ds ratios in genes encoding the PE/PPE proteins occur when genomes of relatively distantly related species such as M. cannettii and M. marinum are used as the reference genome (Tables 6 and 7). In other words, different selection effects are applied to PE/PPE protein genes versus other protein-coding genes in Mycobacterium evolution. Our observation coincides with those of previous studies that suggested a general positive selection or relaxation of negative selection in the molecular evolution of PE/PPE proteins in MTB. 28 We believe that the second explanation is more likely in the case of MTB for the following reasons. (a) The effective population size of MTB has been significantly reduced because of its pathogenic lifecycle. This reduction in population size usually leads to decreased efficiency of selection, thus allowing deleterious mutations to accumulate in the genome. (b) The MTB–M. marinum dn/ds ratios are generally smaller than the ratios between different MTB strains. This is analogous to “smaller between-species distance than within-species distance.” The small (between-species) divergence usually indicates negative selection, whereas the larger (within-species) diversity usually means relaxation of negative selection, unless some type of diversifying selection has been in action.
Diversifying selection can cause remarkable genetic and phenotypic differences between different strains. One good example is the skin color of different human races. However, we will need stronger evidence and good biological explanations to claim this type of selection in MTB. In addition, we would like to emphasize that relaxed negative selection can sometimes be the source of functional innovations. A classical theory is “evolution by duplication,” which means that duplicated genes (as in the case of the PPE family) are subject to relaxed negative selection because of functional redundancy. Therefore, they are free to evolve and may accidentally acquire new functions that may increase the fitness of the carrier organisms. Such functions and the related genes will be subject to positive selection afterward. So the type of selection actually changes over time and with the context of evolution. In contrast, the differing dn/ds ratios between PE/PPE protein genes and all protein-coding genes are not detectable in most of the Taiwan MTB strains when H37Rv or M. africanum is used as the reference. The A18 MTB strain (EAI2_Manilla) is an exception, in that it was the only strain that showed elevated dn/ds values in the PE/PPE family, regardless of the reference genome used for SNP calling. This suggests a different fate for PE/PPE proteins in the evolution of the A18 strain in contrast to the other five strains from Taiwan.
Due to a complex interaction between the host, the pathogen, and the environment, the outcome of MTB infection and disease is highly variable.29,30 There is mounting evidence that this variable outcome may be influenced by MTB genomic diversity.29,31 However, the evolutionary forces that shape this variation are not well understood. Genomic comparisons have identified genetic variation for population screening; however, these analyses are limited to relatively few genetic loci that vary between the compared genomes and therefore are potentially misleading.14,32,33 Nucleotide sequences provide robust data for studying population variation. The mutational processes that generate this variation are understood, and sequence data have been successfully used in the study of bacterial epidemiology, population structure, and evolution. 34 The complete genome sequences14–16,34 provide access to all regions of the chromosome and facilitate such studies.
A previous study of MTB strains in Taiwan by our group (based on ST and VNTR-MIRU analysis) revealed an interesting association of strains with historical migrations of different ethnic populations. 35 Comparing whole-genome sequences of the main MTB strains in Taiwan in the present study confirmed the previous findings, thus establishing a good model to explore the global evolution and spread of MTB. The genome sequences of MTB strains isolated from representatives of Taiwanese aborigines (M3/Haarlem-3, M7/LAM-3, M24/Beijing ancient strain), a representative of a recent Han immigrant (W6/Beijing modern strain), and representatives of historic Han immigrants (A18/EAI2_MANILLA, A27/T1) were determined by 454 sequencing technology. 11 More than 95% of the reads were assembled into sequence contigs. The sequence data from these representative strains will be further analyzed to discover the unique genomic features of MTB infecting different ethnic groups in Taiwan.
At present, we are focusing on MTB genomic information together with epidemiological and clinical data in order to identify factors significant in transmission, virulence, drug resistance, and protection efficacy of vaccines among different strains. We conducted Ka/Ks analysis to identify selection pressure on the protein-coding regions. As shown in Table 7, we found that, in general, there are more N than S changes in the MTB-coding sequences. Second, we found that the M7 strain isolated from an aboriginal patient is most closely related to the F11, an MTB strain isolated from a TB epidemic in the Western Cape of South Africa. This finding further supports our previous study, in which we demonstrated that the Haarlem and LAM lineages were circulating in the aboriginal community in Taiwan and suggesting a link of these strains to post-Columbus Europeans.6,36
The Beijing strains have spread worldwide as a genetically conserved genotype of MTB, often in association with drug resistance.8,37–40 This worldwide spreading in the population structure of MTB is driven in part by man-made factors, and perhaps also linked with intrinsic mycobacterial characteristics. By using DNA MassARRAY® technology (Sequenom), we have established protocols for rapid and cost-effective assays for distinguishing different sublineages of Beijing strains. 33 Moreover, we found SNPs in a putative DNA repair gene, which may be involved in facilitating spreading of the pathogen, but did not demonstrate an association with multidrug resistance.33,41–45 Furthermore, we are conducting informatics analysis of the six sequenced Taiwan MTB genomes. Preliminary data indicate as many as 620 SNPs in at least two of the sequenced strains. The information generated by comparative analysis is the basis for establishing an MTB genotyping procedure for tracing the evolution and distribution of different MTBs in Taiwan. Molecular population genetic analysis of clinical strains delineates relationships among closely related strains of pathogenic microbes and allows construction of genetic frameworks for examining the distribution of biomedically relevant traits, such as virulence, transmissibility, and host range. In seeking to describe the distribution of characteristics of MTB strains and identify the determinants of that distribution, we are attempting to identify factors that determine disease transmission. Comparative genomic hybridization microarray chips will be designed based on the determined genomic sequences in order to conduct population genetic studies quickly and efficiently. Such studies will not only help us to understand the dynamics of TB transmission in Taiwan but will also combine sequence analysis and microarray technology for investigating drug resistance and virulence.
Conclusions
We demonstrated that Haarlem and LAM MTB strains are present in the aboriginal community in Taiwan, suggesting a link of these strains to post-Columbus Europeans. Taken together, our results revealed an interesting association of MTB strains with historical migrations of different ethnic populations, thus providing a good model to explore the global evolution and spread of MTB.
Author Contributions
Conceived and designed the experiments: H-YD, S-FT. Analyzed the data: Y-YC, Y-TC, J-RC, C-HL, K-MW, M-SL. Wrote the first draft of the manuscript: H-YD, Y-YC, Y-TC, C-HL, I-JS, S-FT. Contributed to the writing of the manuscript: H-YD, Y-YC, Y-TC, C-HL, S-FT. Made critical revisions and approved the final version: H-YD, Y-YC, Y-TC, J-RC, C-HL, K-MW, M-SL, I-JS, S-FT. All the authors reviewed and approved the final manuscript.
Footnotes
Acknowledgments
We thank the mycobacteriology laboratories of Mennonite Christian Hospital, Tri-Service General Hospital, and Wan-Ciao Veterans Hospital for providing bacterial isolates. All participants of this consortium are acknowledged for valuable discussions.