
Abstract

Losartan prevents acquired epilepsy via TGF-β signaling suppression
Bar-Klein G, Cacheaux LP, Kamintsky L, Prager O, Weissberg I, Schoknecht K, Cheng P, Kim SY, Wood L, Heinemann U, Kaufer D, Friedman A; Losartan prevents acquired epilepsy via TGF-β signaling suppression. Ann Neurol 2014;75:864–875. doi: 10.1002/ana.24147
OBJECTIVE: Acquired epilepsy is frequently associated with structural lesions after trauma, stroke, and infections. Although seizures are often difficult to treat, there is no clinically applicable strategy to prevent the development of epilepsy in patients at risk. We have recently shown that vascular injury is associated with activation of albumin-mediated transforming growth factor β (TGF-β) signaling, and followed by local inflammatory response and epileptiform activity ex vivo. Here we investigated albumin-mediated TGF-β signaling and tested the efficacy of blocking the TGF-β pathway in preventing epilepsy. METHODS: We addressed the role of TGF-β signaling in epileptogenesis in 2 different rat models of vascular injury, combining in vitro and in vivo biochemical assays, gene expression, and magnetic resonance and direct optical imaging for blood–brain barrier permeability and vascular reactivity. Long-term electrocorticographic recordings were acquired in freely behaving animals. RESULTS: We demonstrate that serum-derived albumin preferentially induces activation of the activin receptor-like kinase 5 pathway of TGF-β receptor I in astrocytes. We further show that the angiotensin II type 1 receptor antagonist, losartan, previously identified as a blocker of peripheral TGF-β signaling, effectively blocks albumin-induced TGF-β activation in the brain. Most importantly, losartan prevents the development of delayed recurrent spontaneous seizures, an effect that persists weeks after drug withdrawal. INTERPRETATION: TGF-β signaling, activated in astrocytes by serum-derived albumin, is involved in epileptogenesis. We propose losartan, a drug approved by the US Food and Drug Administration, as an efficient antiepileptogenic therapy for epilepsy associated with vascular injury.
Commentary
Breaches in the blood brain barrier (BBB) are linked to paroxysmal epileptiform activity and epileptogenesis in acquired epilepsy resulting from head trauma, infection, tumors and other forms of brain injury. Exactly how BBB disruption triggers the development of focal epilepsy is an important area of investigation. Astrocytic mechanisms for controlling spatial potassium ion (K+) buffering, reduced adenosine, and altered metabotropic glutamate receptor-mediated signaling have all been implicated in epileptogenesis, but our understanding of how disrupting the BBB leads to these events remains incomplete. The study discussed here examined how breakdown of the BBB and vascular coupling to astrocytes can lead to changes in downstream signaling pathways in astrocytes. Losartan was tested as a novel drug treatment for preventing epileptogenesis after BBB compromise. This drug appears to knock out astrocyte signaling after vascular damage.
To experimentally induce a lasting and focal breach in the BBB, the authors of this study applied bile salts dehydrocholic acid or deoxycholic acid (DOC) to the cortical surface. Bile salts open the tight junctions in brain capillaries, mimicking focal BBB dysfunction following pathologic events such as head trauma. Because this experimental approach causes inflammation without concomitant neuronal injury, it eliminates neurodegeneration as a confounding variable, allowing more precise analyses of the role of astrocytes in the temporal sequence of cellular and molecular events triggering epileptogenesis. Serum proteins traverse the local gap in the BBB created by topical application of DOC and can be tracked with microscopic, biochemical, or spectrophotometric methods to pinpoint the first cell types to respond to BBB damage and the ensuing chain of events that lead to epileptiform activity.
Astrocytic changes following the disruption of the BBB have become a prime target for therapeutic interventions in focal epilepsy (1). Astrocytic activation is coupled to BBB breakdown and serum albumin uptake into astrocytes. Albumin uptake by astrocytes reduces the expression of inward-rectifying potassium channel, Kir4.1, the predominant potassium channel expressed by astrocytes responsible for their ability to buffer extracellular K+ ions (2). Albumin also reduces astrocytic expression of aquaporin 4, increasing extracellular water (3, 4). Experimentally-triggered focal breakdown of the BBB by DOC, or directly applying albumin to the rat somatosensory and motor cortex through cranial windows, dramatically increases reactive astrocytes within a day, and causes epileptiform activity within 4 days (5). Albumin triggers astrocytes to release the inflammatory cytokine interleukin-1β (IL-1β) and transforming growth factor β1 (TGFβ1), promoting signaling downstream of TGFβ1 receptors and astrocyte proliferation (6).
Changes in astrocytes after albumin uptake contribute to neuronal synchrony and hyper-excitability. IL-1β release by reactive astrocytes reduces seizure thresholds during the period of epileptogenesis in both the kindling and pilocarpine models of temporal lobe epilepsy (4, 7). The central role played by albumin-mediated astrocyte activation in epileptogenesis is further supported by studies in multiple rodent models of epileptogenesis, including the isolated cortex model in vitro (8), chemical kindling (9), and the systemic pilocarpine model. Notably, a hallmark of these models is local astrocyte activation in the focal region of BBB breakdown. After a delay, epileptiform activity develops, along with glutamatergic and GABAergic neurotransmitter receptor alterations, ionic imbalances, and axonal rewiring within cortical circuits.
Although prior work identified serum albumin infiltration across a breach in the BBB as the key precipitating event leading to astrocyte activation in epileptogenesis, the mechanisms for albumin uptake and the ensuing downstream signaling cascades were not well understood. To delineate these pathways, Bar-Klein and colleagues examined the mechanisms for astrocytic serum albumin uptake pharmacologically, blocking caveolae-mediated endocytosis or TGFβ-1 receptor-mediated albumin uptake. Blockade of either or both uptake pathways reduced albumin uptake by cortical astrocytes, but only partially, suggesting the involvement of other undefined pathways. Bar-Klein and colleagues next examined expression of TGFβ and its receptors in astrocytes and showed that albumin uptake upregulated TGFβ1 mRNA and increased secretion of TGFβ1 protein, in a positive feedback loop. TGFβ1 levels are approximately 100-fold greater in astrocytes compared to neurons, and albumin uptake dramatically upregulated them. Albumin uptake in astrocytes also increased the enzymatic activity of the Activin Like Kinases 1 and 5 (ALK 1/5), two protein kinases responsible for phosphorylating intracellular transcription factors downstream of TGFβ receptors.
Having established some of the key events in the signal transduction cascade following albumin uptake in astrocytes, Bar-Klein and co-workers examined the subsequent signals within astrocytes. They focused on ALK 1/5-dependent phosphorylation of the mammalian Smad proteins. The Smads are intracellular signaling intermediaries for the TGFβ growth pathway and act as transcription factors. Albumin-mediated TGFβ signaling in cortical astrocytes leads to Alk5-dependent phosphorylation of the receptor-regulated Smad (R-Smad) family members, Smad2/3. Upon phosphorylation, they form Smad transcription factor complexes at gene regulatory regions (10). A rise in p-Smad 2/3 levels occurs after exposure to albumin in just over a day. A summary diagram of the findings linking blood-vessel leakage of albumin, astrocyte activation, TGFβ1 dependent intracellular signaling pathways, and the development of neuronal paroxysmal epileptiform activity is shown in Figure 1 (reprinted with permission) (11).
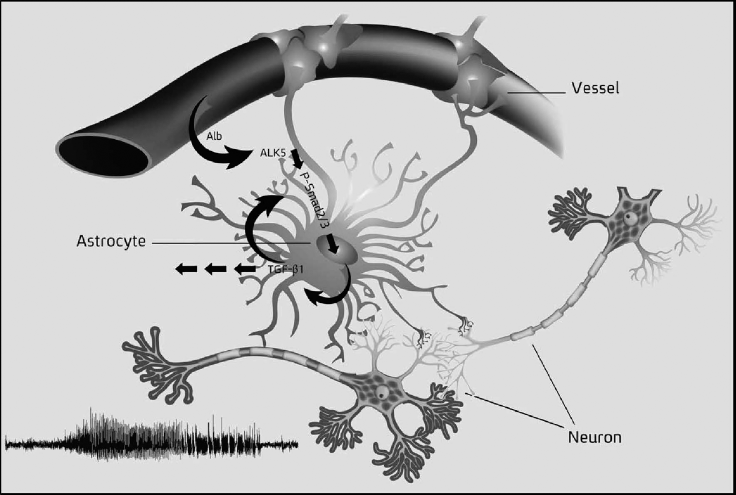
The central role played by astrocytes after focal disruption of the blood brain barrier (BBB). The astrocyte in the center of the figure has attachments to capillaries (Vessel) and neurons at synaptic sites. Breakdown of the BBB allows infusion of albumin (Alb) into the parenchyma, which is taken up by astrocytes. Uptake activates ALK5-dependent phosphorylation of p-Smad 2/3, transcriptional coregulators for target genes, including TGFβ1. Reprinted with permission (11).
The Smad signaling cascade appears to be the tip of the iceberg. Would disrupting this cascade prevent astrocyte activation, cytokine release, and changes in gene expression in astrocytes that contribute to neuronal hyperexcitability and epileptogenesis? If so, this intervention could lead to new treatments to prevent acquired epilepsy after traumatic brain injury. Bar-Klein and colleagues tested these questions with the clinically relevant TGFβ inhibitor losartan. This is an FDA-approved angiotensin II type 1 (AT1) receptor antagonist drug marketed by Merck & Co. Losartan, available under the trade name Cozaar (or as a generic), was found to block TGFβ signaling in peripheral tissue and is also used to treat hypertension. To directly examine the effects of losartan on reactive astrogliosis, Bar-Klein and colleagues installed cranial windows directly over somatosensory and motor cortex to monitor astrocytes and cortical vasculature while infusing albumin, with or without losartan. Remarkably, p-Smad 2/3 phosphorylation was blocked in the presence of losartan. Prior work showed that activated Smad1 forms a complex with the factors p300 and Stat3, which upregulates glial fibrillary acidic protein (GFAP), and directs the proliferation of astrocytes. Consistent with prior results, losartan also blocked GFAP upregulation, indicating that the drug suppressed astrogliosis following exposure to albumin.
The authors also monitored the development of spontaneous epileptiform activity after cortical exposure to albumin in vivo, by means of EEG recordings for up to 110 days, with or without losartan treatment. More than 85 percent of rats with cortical albumin treatment developed spontaneous recurrent seizures, beginning 2 days after the onset of albumin infiltration. The rats had an average of about six seizures per week, with average seizure durations of approximately 14 seconds. Co-infusion of losartan with albumin markedly reduced the percentage of epileptic rats from 85 to 25 percent, and cut down the average seizure duration to just 5 seconds. This is a striking therapeutic effect from just a single dose of losartan applied topically to the cortex.
To examine losartan's efficacy in a more clinically relevant paradigm, the authors next asked whether losartan might prevent acquired epilepsy after focal vascular injury caused by delivering DOC to the cortical surface through a craniotomy window. DOC delivery was followed 40 minutes later by an intraperitoneal injection of losartan. The drug was then supplied to the rats in their drinking water for the duration of the study. While all of the DOC-treated rats developed epileptiform activity, 50 percent fewer developed spontaneous recurrent seizures after receiving losartan.
In summary, BBB breakdown and vascular coupling to astrocytes are linked to development of seizures after traumatic brain injury. The new findings discussed here indicate a central role for TGFβ signaling in triggering reactive astrogliosis following BBB breakdown. The remarkable anti-epileptogenic effect of the drug losartan following vascular injury in rodents, points to losartan as a potential treatment for preventing the development of epilepsy in patients with acute brain injury.