Rivaroxaban and amiodarone are commonly used for treating patients with atrial fibrillation. Drug-drug interactions between rivaroxaban and amiodarone may increase exposure to rivaroxaban. However, the clinical relevance of this drug-drug interaction is still not clear.
Objective:
The aim was to investigate the risk of bleeding in patients receiving a combination of rivaroxaban and amiodarone.
Methods:
This was a prospective observational study in which we included atrial fibrillation patients treated with rivaroxaban. The patients were divided into the rivaroxaban group and the combination of rivaroxaban and amiodarone group (the combination group). Propensity score matching (PSM) and inverse probability of treatment weighting (IPTW) were performed to adjust between-group differences. The primary endpoint was defined as the time to the first occurrence of a composite of major, clinically relevant nonmajor, and minor bleeding.
Results:
In total, 481 atrial fibrillation patients were included in the analysis. After PSM, 154 patients in the rivaroxaban group were matched with 154 patients in the combination group. The bleeding events mainly consisted of clinically relevant nonmajor and minor bleeding. Only one patient experienced major bleeding. The primary outcome was recorded in 26.0% of patients in the combination group and 10.4% of patients in the rivaroxaban group (hazard ratio = 2.76, 95% CI = 1.55-4.93, P < 0.001). The bleeding risk was significantly higher in the combination group compared with that in the rivaroxaban group in the IPTW and stabilized IPTW analyses (hazard ratio = 2.17, 95% CI = 1.32-3.56, P = 0.002).
Conclusion and Relevance:
The combination of rivaroxaban and amiodarone increased the risk of bleeding in patients with atrial fibrillation, especially clinically relevant nonmajor and minor bleeding. Physicians prescribing rivaroxaban and amiodarone together should be concerned about an increase in the risk of nonmajor bleeding.
Research article
Restricted accessResearch articleFirst published August, 2024pp. 771-780
Virginia H. FlemingORCID, Jianing Xu, Xianyan Chen , [...]
View All
Abstract
Background:
Fluoroquinolones (FQs) are associated with increased risk of tendon injury but comparative risk versus other antibiotic options for the same indication has yet to be evaluated.
Objective:
Describe the incidence (relative risk) of any tendon injury in patients receiving FQ compared with other (non-FQs) antibiotics for treatment of community-acquired pneumonia (CAP).
Methods:
A retrospective propensity score weighted cohort study was performed to evaluate the association between FQ antibiotics and tendon injury risk at 2 time points (within 1 month and within 6 months of use) compared with non-FQ regimens for treatment of CAP. The evaluation was performed using the CCAE (MarketScan Commercial Claims and Encounters) and COB (Medicare Supplemental and Coordination of Benefits) databases from 2014 to 2020. Patients with ICD (International Classification of Diseases) 9/10 coding for outpatient pneumonia who were >18 years and without history of tendon injury were included. Patients with history of tendon injury, who received multiple antibiotic therapies for recurrent pneumonia, or who received both FQ and non-FQ regimens during the study period were excluded. Propensity score weighting was used to adjust for selection bias due to contributing risk factors, including demographics (age, sex), comorbidities (diabetes mellitus, chronic kidney disease), and concurrent medications (corticosteroids).
Results:
At 1 month, the odds of tendon injury were estimated to be significantly higher (41.9%) in patients receiving FQs compared with those receiving a non-FQ-based regimen (odds ratio [OR] = 1.419, 95% confidence interval [CI] = [1.188-1.698]). The odds of tendon injury were also estimated to be higher (OR = 1.067, 95% CI = [0.975-1.173]) in the FQ population within 180 days, but this effect was not statistically significant. The most frequent sites of tendon injuries were rotator cuff, shoulder, and patellar tendon.
Conclusions and Relevance:
Prescribers should recognize the risk of tendon injury within 1 month of FQ use when considering treatment regimens for CAP and use alternative options with lower risk whenever possible.
Research article
Restricted accessResearch articleFirst published August, 2024pp. 781-789
Courtney S. Pilkerton, Megan Adelman, Emily Crocetti , [...]
View All
Abstract
Background:
Direct-acting oral anticoagulants (DOACs) have become the preferred drugs for managing venous thromboembolism (VTE). Despite their advantages over vitamin K antagonists such as warfarin, their use in obese patients remains controversial with many providers reluctant to switch patients managed on warfarin. Outcome research that opts to increase provider confidence when prescribing DOACs for patients with obesity will be invaluable.
Objective:
This investigation evaluated whether patients with a body mass index (BMI) 35 kg/m2 or greater who were prescribed a DOAC had a higher risk for a recurrent VTE or bleed event relative to warfarin.
Methods:
The study was conducted in West Virginia which has the highest rate of obesity in the United States.
Results:
Of the total study population (1633), 2.3% (37) had a recurrent thrombotic event, 5.5% (89) had a major bleed event, and 10.7% (174) had some type of bleeding event. No individual patient characteristic was associated with recurrent thrombosis—including BMI. Older age, antiplatelet use, and taking a medication with a theoretical risk of increasing the effect of DOACs were associated with any and major bleeding events. The use of warfarin was associated with major bleeding events more frequently versus a DOAC. Body mass index was not a predictor for recurrent VTE or any bleed or major bleed events.
Conclusions:
These findings support the conclusion that DOACs are an appropriate and effective drug class for the management of VTE in patients with obesity.
Research article
Restricted accessResearch articleFirst published August, 2024pp. 790-795
Patiromer and sodium zirconium cyclosilicate (SZC) are 2 oral potassium binders approved for chronic hyperkalemia. It is unknown if one is more effective at reducing serum potassium than the other in acute hyperkalemia.
Objective:
The objective of this study was to determine if there was a difference between patiromer and SZC in the reduction of serum potassium in patients with acute hyperkalemia.
Methods:
This was a single-center, retrospective, observational study. Patients with a nonhemolyzed serum potassium level of 5.5 mEq/L or greater and received at least one dose of patiromer or SZC were included. The primary outcome was to determine the difference in effectiveness between patiromer and SZC in lowering of serum potassium 6 to 24 hours after administration. Secondary outcomes included description of total dosage received in 24 hours and incidence of electrolyte changes.
Results:
A total of 200 patients were included in this study, with 100 patients in each group. Serum potassium was significantly reduced by both patiromer (−1.2 mEq/L, 95% confidence interval [CI]: −2.3 to −0.2) and SZC (−0.8 mEq/L, 95% CI: −1.0 to −0.7), but there was no difference between the 2 medications in the amount of potassium reduction (P = 0.464). No clinically significant differences in electrolyte changes were seen.
Conclusions and Relevance:
This study represents the first head-to-head comparison of patiromer and SZC in the setting of acute hyperkalemia. No difference in effectiveness between patiromer and SZC in reducing serum potassium was seen. Both agents can be considered in acute hyperkalemia management.
Research article
Restricted accessResearch articleFirst published August, 2024pp. 796-802
Jillian L. DescourouezORCID, Jeannina A. Smith, Christopher M. Saddler , [...]
View All
Abstract
Background:
Cytomegalovirus (CMV)-specific cell-mediated immunity is important for control of CMV after transplant. Assays exist to measure this, but their place in therapy is unclear, particularly in CMV high-risk recipients, without pretransplant exposure.
Objective:
The objective of this study was to evaluate predictive potential of a positive assay to determine freedom from DNAemia and describe subsequent 3-month CMV outcomes.
Methods:
Adult CMV high-risk kidney and/or pancreas transplant recipients were included if a CMV inSIGHT T Cell Immunity Panel (TCIP, Eurofins Viracor) was ordered and resulted between 1 August, 2019 and 30 July, 2022.
Results:
Seventy-six patients were included in our study; 49 tested during prophylaxis and 27 during treatment. Most TCIP assays obtained in the prophylaxis cohort were negative (n = 46, 93.9%). Rate of post-TCIP CMV infection was 10.2%. In those tested during treatment, 33.3% were positive and rate of post-TCIP CMV recurrence was 22.2%. The positive predictive value of the assay to successfully predict immunity was 66.7% during both prophylaxis and treatment. There were 4 cases of TCIP predictive failure with progressive CMV replication. At time of replication, 2 patients had concomitant clinical confounders thought to influence immune control of viral replication. All patients had intensification of immunosuppression prior to recurrent replication, but after TCIP was collected.
Conclusion and Relevance:
The data obtained from the TCIP are not static, immune control of CMV in latency can change and must be evaluated in clinical context. Timing of TCIP after transplant is significant, and patient-specific factors remain important to assess the likelihood of CMV in each unique patient-specific scenario. A CMV stewardship program can aid in application and interpretation of results.
Research article
Restricted accessResearch articleFirst published August, 2024pp. 803-810
Madeline MitchellORCID, Danine Sullinger, Duke Dyer , [...]
View All
Abstract
Background:
Patients with cardiogenic shock or end-stage heart failure can be maintained on mechanical circulatory support (MCS) devices. Once a patient undergoes placement of a device, obtaining and maintaining therapeutic anticoagulation is vital. Guidelines recommend the use of institutional protocols to assist in dosing and titration of anticoagulants.
Objective:
The purpose of this study was to characterize the use of bivalirudin before and after the implementation of a standardized titration protocol in patients with MCS.
Methods:
A retrospective review of patients who received bivalirudin for MCS (VA ECMO [veno-arterial extracorporeal membrane oxygenation], Impella, or LVAD [left ventricular assist device]) before and after the implementation of the titration protocol into the electronic health record (EHR) was conducted. The primary outcome was to compare the proportion of therapeutic activated partial thromboplastin time (aPTT). Secondary outcomes included number of subtherapeutic and supratherapeutic aPTTs, incidence of bleeding and clotting events, bivalirudin titrations per day, and percentage of patients with therapeutic aPTT level.
Results:
A total of 100 patients were included (precohort = 67; postcohort = 33). The proportion of therapeutic aPTTs was significantly higher in the postcohort than that in the precohort (62% vs 48%; P < 0.001). The postcohort had 0% of patients failing to achieve therapeutic aPTT levels. The number of titrations per day was significantly lower in the postcohort, with 1.20 titrations per day versus 1.93 in the precohort (P < 0.001).
Conclusions:
Implementation of the bivalirudin titration nomograms within the EHR significantly increased the number of therapeutic aPTTs, reduced the number of patients who never achieved a therapeutic aPTT, and reduced the required number of titrations per day.
Review article
Restricted accessReview articleFirst published August, 2024pp. 811-826
The objective of this systematic review is to determine the tolerability and safety of psilocybin in a variety of psychiatric and substance-dependence conditions.
Data sources:
A systematic review was conducted using Embase, PubMed, Cochrane Central, and Web of Science through September 2023 using the following terminology: “psilocybin” AND “mental-disease” OR “substance-dependence” AND “disease-therapy,” in addition to other synonymous key words.
Study selection and data extraction:
Literature reporting acute effects and safety data following the use of psilocybin as the pharmacologic intervention in a clinical trial in adult patients with a psychiatric or substance-dependence condition were included. Following the application of inclusion and exclusion criteria, 16 studies were ultimately included in this review.
Data synthesis:
The most common treatment-emergent adverse effects reported were transient nausea and headache. Transient anxiety was reported as a frequent psychiatric effect, and 3 participants received a benzodiazepine for refractory anxiety during the psilocybin session. Psilocybin demonstrated modest increases in blood pressure and heart rate, and 1 participant received an antihypertensive for sustained hypertension during the psilocybin session. No cases of psilocybin-induced psychosis or Hallucinogen Persisting Perception Disorder were reported.
Relevance to patient care and clinical practice:
Treatment resistance remains a concern for psychiatric patients and novel therapies are needed to help alleviate the burden of morbidity and mortality. Psilocybin demonstrates promising acute and long-term safety that may allow for its use in psychiatric or substance-dependence conditions as an alternative to standards of care or in treatment-resistant patients.
Conclusions:
Psilocybin has demonstrated tolerability and safety in recent literature that has investigated its therapeutic potential in a variety of psychiatric or substance-dependence conditions.
Review article
Restricted accessReview articleFirst published August, 2024pp. 827-833
Amber Lanae MartirosovORCID, Christopher GiulianoORCID, Macy Shupp , [...]
View All
Abstract
Objective:
The objective is to review the pharmacology, efficacy, and safety of intranasal zavegepant in the acute treatment of migraine with or without aura.
Data source:
PubMed, Embase database, and ClinicalTrials.gov were searched using the following terms: Zavzpret, Zavegepant, BHV-3500, and migraine.
Study selection and data extraction:
Articles published in English from January 2013 to September 2023 related to pharmacology, safety, efficacy, and clinical trials were assessed.
Data synthesis:
In a phase 2/3 trial, zavegepant 10 and 20 mg were more effective than placebo on primary endpoints of freedom of pain (22.5%, 23.1%, and 15.5%, respectively), and freedom from most bothersome symptoms (MBSs) (41.9%, 47.9%, and 33.7%, respectively) 2 hours after treatment. The incidence of adverse effects for both doses was similar to placebo. In a phase 3 trial, zavegepant 10 mg was compared with placebo. Two hours after treatment, more patients in the zavegepant group achieved pain freedom (24% vs 15%) and relief from MBSs (40% vs 31%) compared with placebo. Common adverse events included dysgeusia (21% zavegepant vs 5% placebo) and nasal discomfort (5% zavegepant vs 1% placebo).
Relevance to patient care and clinical practice in comparison with existing drugs:
Zavegepant is indicated for acute treatment of migraine with or without aura in adults. Zavegepant method of administration and prompt relief of migraine symptoms may be an attractive alternative to triptans for those in need of relief.
Conclusion:
Zavegepant may be a convenient and useful acute treatment option for migraines with and without aura.
Review article
Restricted accessReview articleFirst published August, 2024pp. 834-848
To review efficacy and safety data of valoctocogene roxaparvovec (Roctavian) and etranacogene dezaparavovec (Hemgenix), novel gene therapies for the treatment of the life-threatening bleeding disorders hemophilia A and B, respectively.
Data Sources:
A PubMed/Google Scholar search from inception through August 11, 2023 was conducted using the following keywords: gene therapy, hemophilia A, hemophilia B, etranacogene dezaparavovec, valoctocogene roxaparvovec, and bleeding.
Study Selection and Data Extraction:
Data, including phase 1 to 3 clinical trials (non-comparator), were obtained from primary literature and package inserts. These reports evaluated clinical pharmacology, efficacy, safety, adverse events, warnings, and precautions.
Data Synthesis:
Valoctocogene phase 3 study in males (n = 134): 87% had factor VIII (FVIII) levels that at least met criteria for mild hemophilia. Etranacogene phase 3 study in males (n = 54): within 3 weeks of infusion, mean factor IX (FIX) levels had reached 26.8 IU/dL. Both therapies provided clinically and statistically significant decreases in bleeding events and prophylactic factor infusions. Most common adverse event was elevations in liver function tests that were treated with glucocorticoids.
Relevance to Patient Care and Clinical Practice in Comparison with Existing Drugs:
The endogenous production of clotting factors mimics physiological production while decreasing morbidity and mortality related to bleeding events similar to the effects of existing replacement strategies. Gene therapy was also shown to increase patient quality of life.
Conclusion:
Valoctocogene and etranacogene provide another treatment for selected patients with hemophilia. Treatment for the patient with hemophilia (gene therapy vs replacement strategy) must be personalized as new clinical data are published being cognizant of drug affordability.
Review article
Restricted accessReview articleFirst published August, 2024pp. 849-856
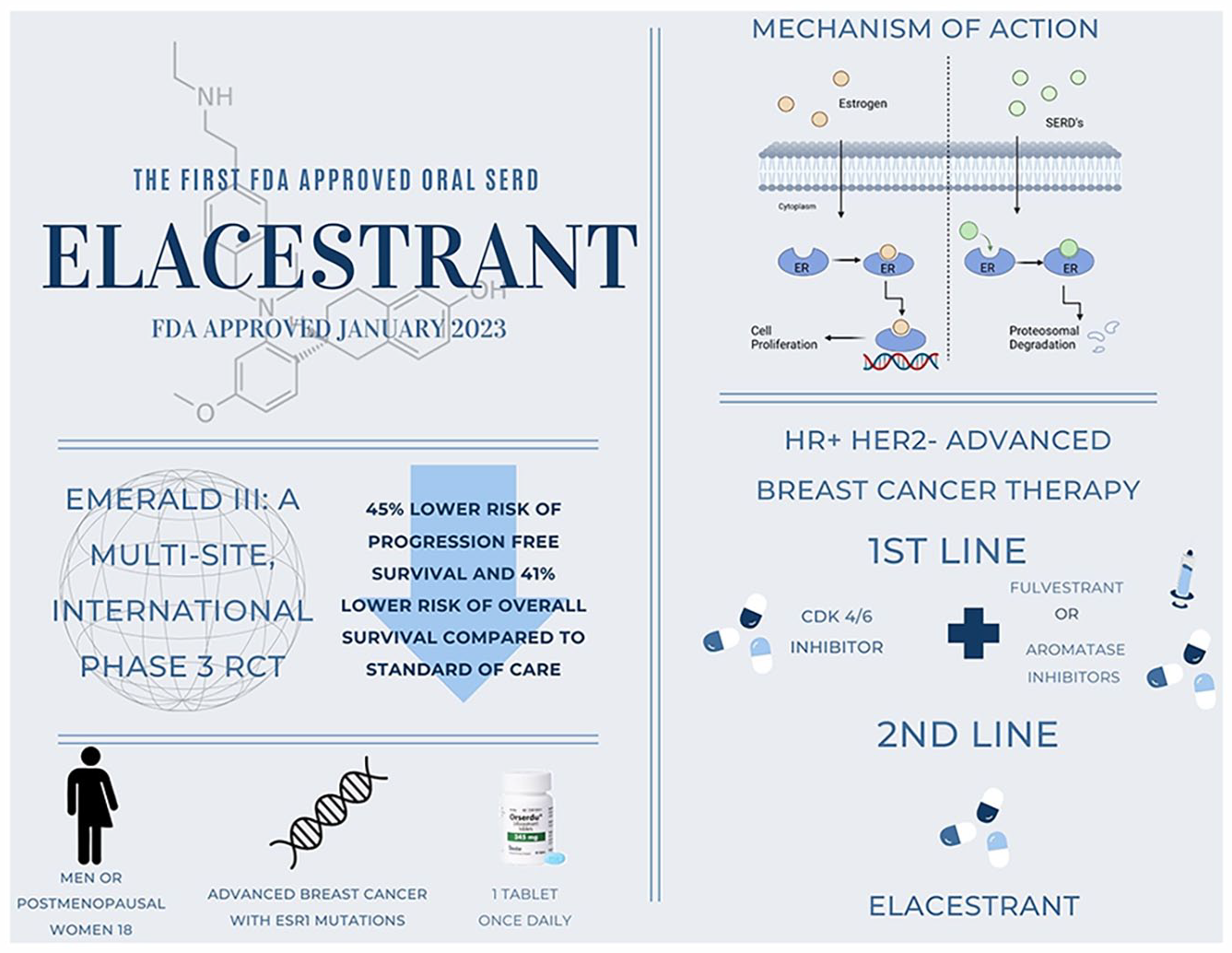
This article aims to discuss elacestrant, an oral selective estrogen receptor downregulator approved by the Food and Drug Administration (FDA) in January 2023 for the treatment of hormone receptor positive (HR+) human epidermal growth factor receptor 2 negative (HER2−) advanced breast cancer.
Data sources:
PubMed, Embase, Medline, Clinicaltrials.gov, and the National Comprehensive Cancer Network (NCCN) were searched from inception to August 31, 2023.
Study selection and data extraction:
Clinical trials published in English were included and relevant information regarding methodology and results were extracted.
Data synthesis:
Phase 1 and 3 trials showed elacestrant was safe and improved progression-free survival in patients with endocrine receptor 1 (ESR1) mutations who failed cyclin-dependent kinase 4/6 inhibitor (CDK 4/6i) plus 1 prior endocrine therapy compared with standard of care (SOC) (fulvestrant, anastrozole, letrozole, or exemestane monotherapy).
Relevance to patient care and clinical practice in comparison to existing drugs:
Elacestrant maintains a comparable adverse event profile with other endocrine therapies and offers an alternative to typical sequential therapy which can delay the use of or be used after traditional chemotherapy. Elacestrant is currently being studied in CDK 4/6 inhibitor naïve patients and as a component of combination therapy for first-line use which could lead to future indications.
Conclusions:
Elacestrant gained FDA approval in January 2023 and can be considered in patients with HR+ HER2− advanced breast cancer and ESR1 mutations who have progressed despite therapy with either CDK 4/6i plus aromatase inhibitors (AI) or fulvestrant or chemotherapy.
Review article
Restricted accessReview articleFirst published August, 2024pp. 857-869
Mrinmayee JoshiORCID, Brendan ClarkORCID, Todd A. Lee
Abstract
Objective:
Several cases of Fanconi syndrome (FS), a severe form of nephrotoxicity, have been reported in patients with HIV on tenofovir-containing antiretroviral therapy. A systematic review of the published literature on tenofovir-related FS in patients with HIV was conducted.
Data Sources:
PubMed and Embase were queried to identify articles in English published between January 2005 and June 2023, reporting tenofovir-related FS in adults with HIV. Preclinical studies, conference/poster abstracts, commentaries and responses, and review papers were excluded.
Study Selection and Data Extraction:
Of the 256 articles screened, 57 met the inclusion criteria. These comprised 37 case reports, 11 case series, 1 cross-sectional study, 1 case-control study, 4 cohort studies, 1 single-arm open-label clinical trial, 1 sub-analysis of clinical trials, and 1 pooled analysis of clinical trials.
Data Synthesis:
Among 56 cases on which information was abstracted, median age at FS diagnosis was 50 years, 51.8% were men, and duration of tenofovir use ranged from 6 weeks to 11 years. Ritonavir was co-prescribed in almost half the cases. In observational and interventional studies, incidence of FS was low. Many studies reported resolution of FS symptoms after tenofovir discontinuation. All FS occurrences were identified in those on tenofovir disoproxil fumarate (TDF), except for one patient on tenofovir alafenamide (TAF).
Relevance to Patient Care and Clinical Practice:
Continuous monitoring of signs and symptoms of renal and bone toxicity is essential for patients with HIV on tenofovir-containing therapy.
Conclusions:
Occurrence of FS is low in patients with HIV treated with tenofovir-based regimens. Concomitant use of ritonavir may increase risk of FS. TAF may be a safer alternative than TDF in terms of nephrotoxicity.
Letter
Restricted accessLetterFirst published August, 2024pp. 870-871