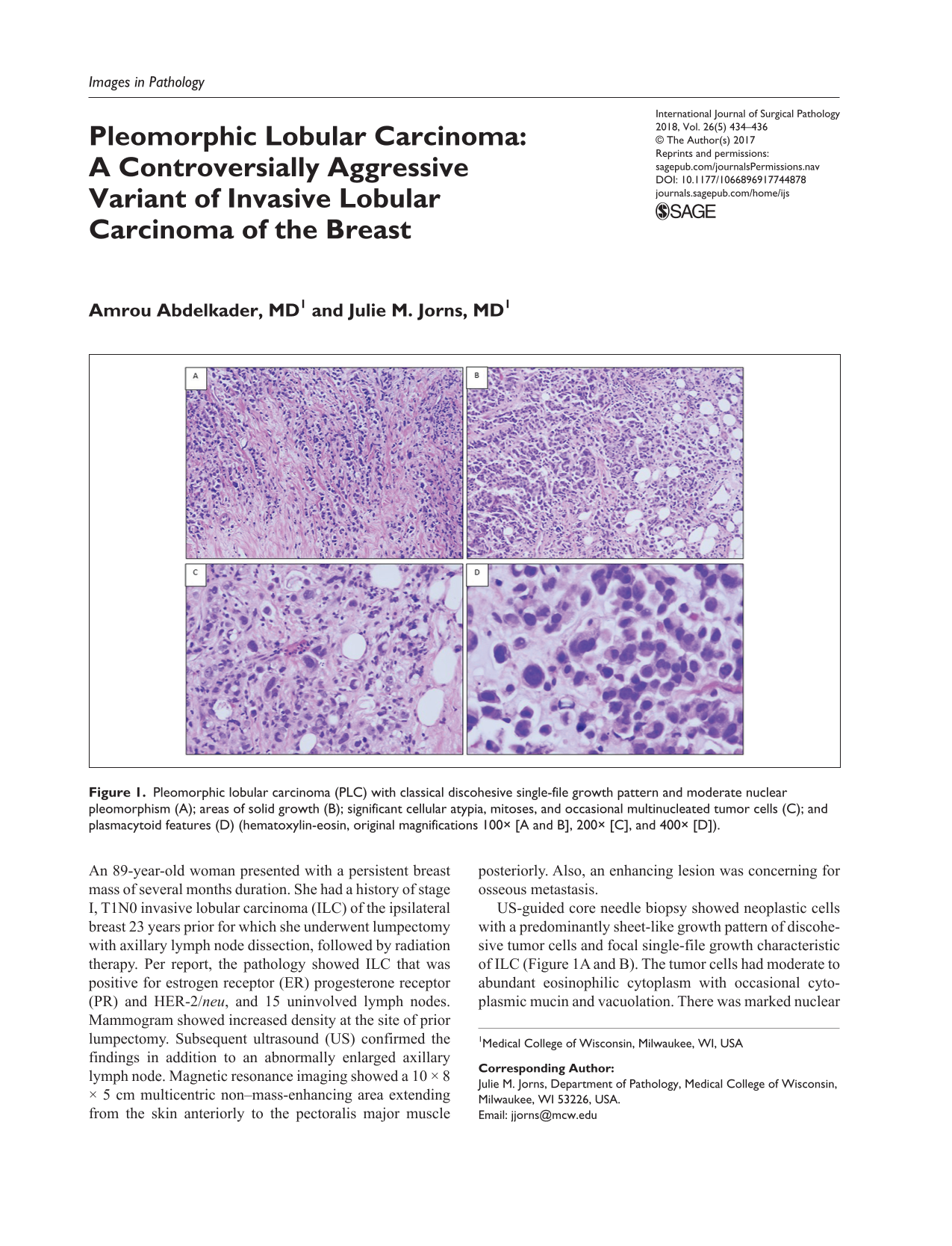
Background. To avoid diagnostic errors such as missed diagnosis and errors in staging tumors due to inadequate tissue sampling, pathologists submit additional sections (AS). Objective. This study assessed frequency, diagnostic yield, distribution, and cost of AS. Method. Among 1542 AS cases, we calculated mean AS per case; fraction of AS that altered diagnosis or stage; AS variation by tissue, malignant versus benign lesions, presence or absence of neoadjuvant therapy, mass, margin, lymph nodes, or other source, resident versus pathologist assistant (PA) dissector; and AS cost per case. Results. Overall 9.2 ± 8.8 AS were collected per case. In only 3.8% (58/1542) of cases AS altered diagnosis or stage. Urinary bladder cases provoked the most AS: 19.5 ± 15.1 per case. Significantly more AS came from malignant versus benign lesions (10.8 ± 9.7 vs 7.6 ± 7.5, P = <.0001) and from specimens treated with neoadjuvant therapy versus malignant lesions not so treated (12.3 ± 9.4 vs 10.3 ± 9.8, P = .02). Lymph nodes were sampled more heavily compared with mass, margin, and other sites combined (11.8 ± 11.4 vs 8.9 ± 8.4, P = .003), but in 78.4% (1209/1542) of cases, AS were from mass. Of diagnosis or stage altering AS cases, two thirds (38/58) were from masses, one fifth (11/58) from lymph nodes, a 10th (6/58) from margins, and a 20th (3/58) from other specimen sites. Resident versus pathologist assistant dissection caused no significant AS difference. AS contributed 40% cost per case. Conclusions. AS per case ranged widely; their diagnostic yield was low; they were highest in urinary bladder specimens, in malignant and particularly neoadjuvant-treated lesions. Although lymph nodes were most heavily sampled, most AS were from masses. Resident dissection did not increase AS and cost of AS was high.