Leuchowius K-J, Jarvius M, Wickström M, Rickardson L, Landegren U, Larsson R, Söderberg O, Fryknäs M, Jarvius J. High content screening for inhibitors of protein interactions and post-translational modifications in primary cells by proximity ligation.Mol Cell Proteomics2010;9:178–183.
Abstract: The cost of developing new drugs is a major obstacle for pharmaceutical companies and academia with many drugs identified in the drug discovery process failing approval for clinical use due to lack of intended effect or because of severe side effects. Since the early 1990s, high throughput screening of drug compounds has increased enormously in capacity but has not resulted in a higher success rate of the identified drugs. Thus, there is a need for methods that can identify biologically relevant compounds and more accurately predict in vivo effects early in the drug discovery process. To address this, we developed a proximity ligation-based assay for high content screening of drug effects on signaling pathways. As a proof of concept, we used the assay to screen through a library of previously identified kinase inhibitors, including six clinically used tyrosine kinase inhibitors, to identify compounds that inhibited the platelet-derived growth factor (PDGF) receptor β signaling pathway in stimulated primary human fibroblasts. Thirteen of the 80 compounds were identified as hits, and the dose responses of these compounds were measured. The assay exhibited a very high Z′ factor (0.71) and signal to noise ratio (11.7), demonstrating excellent ability to identify compounds interfering with the specific signaling event. A comparison with regular immunofluorescence detection of phosphorylated PDGF receptor demonstrated a far superior ability by the in situ proximity ligation assay to reveal inhibition of receptor phosphorylation. In addition, inhibitor-induced perturbation of protein-protein interactions of the PDGF signaling pathway could be quantified, further demonstrating the usefulness of the assay in drug discovery.
Commentary:High content screening (HCS) assays often require the use of antibodies or highly engineered cell lines in addition to fluorescent reporters for detection of signaling events. Immunofluorescence (IF)-based protocols have been used, but these methods suffer from low sensitivity and background fluorescence that lowers the quality of the assay. Assaying for protein–protein interactions (PPIs) in cellular systems is an attractive application of HCS given the low sensitivity of biochemically based PPI assays (e.g., assays based on the binding of isolated polypeptides). This article describes an antibody-based technology that enables HCS of PPIs in primary cells. Some issues related to antibody-based protocols are addressed by employing a signal amplification step based on the ligation of oligonucleotides attached to the antibodies. In the assay, when two antibodies are in close proximity, the oligonucleotides ligate and form a DNA circle. The DNA circle is then replicated by the addition of Φ29 polymerase through a rolling circle amplification (RCA) mechanism with one of the antibody-attached oligonucleotides acting as a primer. The product of the RCA is single-stranded DNA representing copies of the circle to which fluorescently labeled complementary oligonucleotides can bind (see
figure
), resulting in bright sub-micrometer spots at the binding event. The advantages of this proximity ligation assay (PLA) relative to IF techniques is demonstrated using antibodies to the platelet-derived tyrosine kinase growth factor receptor, PDGFRβ, which undergoes phosphorylation and dimerization upon activation. The PLA was designed using antibodies to PDGFRβ and the phosphorylated PDGFRβ. A 96-well plate assay was constructed for both the PLA and IF formats and used to screen a library of 86 kinase inhibitors. The PLA assay showed acceptable performance with a Z′ factor of 0.71, while the IF assay showed unacceptable performance with Z′ factors of −3.1 and 0.25 using either PDGFRβ phosphospecific antibodies or pan-phosphotyrosine antibodies. The increased signal to background ratio for the PLA assay, due to the quantification of rolling circle products instead of total fluorescent intensity, yielded superior sensitivity and assay performance. Employing primary cells in the PLA format eliminated complications related to examining PPIs in engineered cell lines. The PLA technology represents a form of signal amplification technologies that can provide superior sensitivity compared to the use of primary/secondary antibody pairs. The use of appropriately paired antibodies should allow additional protein modification/interaction assays to be adapted to the PLA format. One issue that remains for large-scale screening is the cost of antibodies in the assay, but for focused screening this could be a viable approach for studying PPI intracellularly. Contributed by Doug Auld.
The principle of in situ proximity ligation assay. (a) Phosphorylated PDGFRβ was recognized by a rabbit monoclonal antibody binding the receptor and a pan-specific mouse monoclonal antibody binding phosphotyrosine residues. The primary antibodies were bound by species-specific antibodies conjugated to oligonucleotides. When in proximity, the oligonucleotides could be used as templates for the joining of two additional linear oligonucleotides into a DNA circle. One of the antibody-attached oligonucleotides was extended by rolling circle amplification using the DNA circle as a template to generate a submicrometer-sized bundle of DNA. (b) By hybridizing fluorophore-labeled oligonucleotides to the repeated sequences of the amplification product, the antibody binding event could be visualized by microscopy as bright fluorescent spots. (c) Typical results from the detection of phosphorylated PDGFRβ in primary human fibroblasts. Red indicates in situ PLA signals, green indicates cytoplasmic staining, and blue indicates cell nuclei.
Super Artifacts
Sotoca AM, Bovee TFH, Brand W, Velikova N, Boeren S, Murk AJ, Vervoort J, Rietjens IMCM. Superinduction of estrogen receptor mediated gene expression in luciferase based reporter gene assays is mediated by a post-transcriptional mechanism.J Steroid Biochem Mol Biol2010;122:204–211.
Abstract: Several estrogenic compounds including the isoflavonoid genistein have been reported to induce a higher maximal response than the natural estrogen 17β-estradiol in in vitro luciferase based reporter gene bioassays for testing estrogenicity. The phenomenon has been referred to as superinduction. The mechanism underlying this effect and thus also its biological relevance remain to be elucidated. In the present study several hypotheses for the possible mechanisms underlying this superinduction were investigated using genistein as the model compound. These hypotheses included (i) a non-estrogen receptor (ER)-mediated mechanism, (ii) a role for an ER activating genistein metabolite with higher ER inducing activity than genistein itself, and (iii) a post-transcriptional mechanism that is not biologically relevant but specific for the luciferase based reporter gene assays. The data presented in this study indicate that induction and also superinduction of the reporter gene is ER-mediated, and that superinduction by genistein could be ascribed to stabilization of the firefly luciferase reporter enzyme increasing the bioluminescent signal during the cell-based assay. This indicates that the phenomenon of superinduction may not be biologically relevant but may rather represent a post-transcriptional effect on enzyme stability.
Commentary:Proper interpretation of signal responses derived from assays requires exploring their origins to determine biological relevance. This article presents a detailed investigation into “superinduction,” which refers to compounds that appear more efficacious than natural effectors. The effect that certain isoflavonoids such as genistein have on estrogen receptor (ER)-mediated reporter induction is examined here. When cells that express firefly luciferase (FLuc) in response to the estrogen response elements are treated with genistein, a biphasic concentration–response curve (CRC) is observed (see
figure
). The first phase (10 to 200 nM compound concentration) of the biphasic curve shows an upper asymptote that is close to 100% of estradiol (E2)-treated cells, but the second phase (1 to 5 μM) shows superinduction (∼188% of E2). Several mechanisms could explain superinduction including biologically relevant mechanisms such as up-regulation of cellular ER levels and increased mRNA levels. Alternatively, superinduction could be due to a biologically irrelevant mechanism related to the FLuc reporter enzyme. For this latter mechanism, FLuc levels may increase in a transcriptional and translational independent manner due to compounds that act as FLuc inhibitors that stabilize the reporter protein intracellularly. This leads to an artificial rise in enzyme levels that can be detected as an activation response. Several orthogonal and secondary assays are described here to examine the biological relevance of genistein superinduction. To examine if genistein superinduction of ER activity is cell-type specific, two orthogonal FLuc cell-based assays were used that differed only in the source of ERα. In one case, the cells were engineered to express ERα, while in the other ERα expression was regulated from the endogenous gene. The biphasic CRC was observed in both cell lines, which supports genistein-mediated superinduction not being due to due mechanisms related to ERα expression. In a secondary assay for proliferation (a biologically relevant assay for estrogenic activity; detection did not involve FLuc), no “superproliferation” was observed. As well, assays that used a reporter other than FLuc (green fluorescent protein and β-galactosidase) responded to E2 but failed to show superinduction by genistein. Reverse-transcription polymerase chain reaction (RT-PCR) did not show any increased mRNA production over E2 treatment, suggesting superinduction was not due to transcript production/stabilization. Another possibility tested in this study was if a metabolite of genistein, instead of genistein itself, led to the superinduction. Although a polar metabolite of genistein known as orobol (5,7,3',4'-tetrahydroxyisoflavone) was found in genistein-treated cells, orobol was found to show only the weaker CRC associated with superinduction (and was not biphasic) in ER-mediated FLuc reporter assays. Finally the authors tested to determine if genistein was a direct inhibitor of the FLuc enzyme, which could lead to reporter enzyme stabilization and superinduction related to E2. The isoflavonoid resveratrol, E2, and genistein were tested as inhibitors of FLuc and only resveratrol and genistein showed inhibition of FLuc at concentrations >1 μM. Taken together, these data suggest that the superinduction phenomenon observed for isoflavonoids such as genistein is due to inhibitor-based reporter stabilization. Superinduction observed in FLuc assays may not be due to a biologically relevant mechanism. This article illustrates the critical thinking and assay portfolio required to judge the biological relevance of compounds derived from reporter assays. Contributed by Doug Auld.
Induction of ER-mediated luciferase activity in the ER-T47D-Luc cells upon exposure to various (anti)estrogens. (Anti)estrogens tested include E2 (▪), E2+ 18 nM RU58668 (♦), genistein (∙), genistein + 18 nM RU58668 (○), and genistein + 180 nM RU58668 (×). Induction is expressed relative to maximal E2 response set at 100%. Data points represent the mean of triplicate exposure ± standard deviation. The dashed line indicates from what concentration onwards genistein concentrations superinduction is observed.
Dealing with FLT Interferences
Gakamsky DM, Dennis RB, Smith SD. Use of Fluorescence Lifetime Technology to Provide Efficient Protection from False Hits in Screening Applications.Anal. Biochem.2010; doi:10.1016/j.ab.2010.10.017.
Abstract: This article describes novel data analysis of fluorescence lifetime-based protein kinase assays to identify and correct for compound interference in several practical cases. This ability, together with inherent advantages of fluorescence lifetime technology (FLT) as a homogeneous, antibody-free format independent of sample concentration, volume, excitation intensity, and geometry, makes fluorescence lifetime a practical alternative to the established “gold standards” of radiometric and mobility shift (Caliper) assays. The analysis is based on a photochemical model that sets constraints on the values of fluorescence lifetimes in the time responses of the assay. The addition of an exponential component with free floating lifetime to the constrained model, in which the lifetimes are constants predetermined from control measurements and the preexponential coefficients are “floating” parameters, allows the relative concentration of phosphorylated and nonphosphorylated substrates to be calculated even in the presence of compound fluorescence. The method is exemplified using both simulated data and experimental results measured from mixtures of dye-labeled phosphorylated and nonphosphorylated kinase substrates. A change of the fluorescence lifetime is achieved by the phosphorylated substrate-specific interaction with a bifunctional ligand, where one binding site interacts with the phosphate group and the other interacts with the dye.
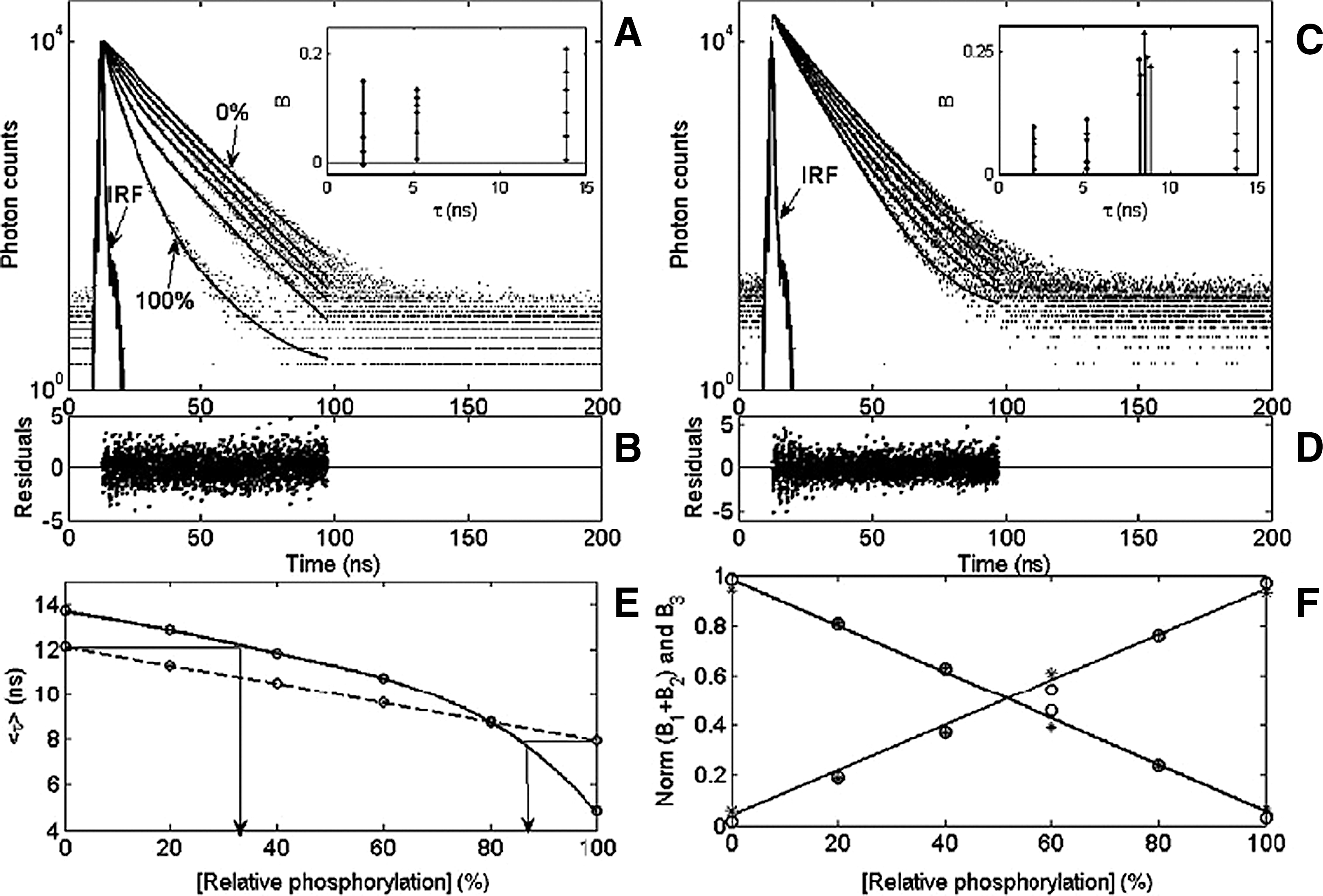
Commentary:Compound fluorescence can interfere with the interpretation of high-throughput screening (HTS) results. Although techniques such as prereading assay plates (e.g., before the initiation of the assay) for compound fluorescence can help flag fluorescent compounds, fluorescence from compound samples can mask the identification of genuinely active compounds. As well, absorbance of excitation or emission light can also complicate the interpretation of results. Fluorescent impurities in compound samples and the conditional nature of fluorescent intensity signals (e.g., fluorophore concentration, excitation wavelength/intensity, and chosen emission wavelength) make it difficult to predict compound fluorescence. Still, fluorescent assays remain one of the most accessible techniques for the development of HTS assays. Techniques to reduce compound interference in fluorescent assays such as the use of red-shifted fluorophores, time-resolved fluorescence and the subject of this article—fluorescent lifetime technology (FLT)—are an area of intense investigation. In FLT, the time a fluorophore spends in the excited state (τ) is measured, which is a property that does not depend on the fluorescent intensity of concentration but instead is modulated by the electronic structure of the fluorophore and the rates of radiative and nonradiative decay processes leading to depopulation of the excited state. Although FLT is insensitive to some modes of interference, it can be affected by collision quenching and background fluorescence of compounds. In this article the authors developed a model to help flag fluorescent interferences in FLT measurements. A protein kinase assay is examined in which the lifetimes of a fluorescently labeled peptide changes when phosphorylated. The assay lifetimes (in this case τ for the phosphorylated and unphosphorylated peptide) are predetermined and a linear model is fitted to the data for which these lifetimes are held constant. A chi-square test can be used to judge the model fits, and an additional lifetime, whose value is allowed to vary in the equation, is used to represent the interfering component in the assay. Experimentally, the model was tested by using an assay for PKBα, in which the lifetime for the fluorescent peptide substrate was modulated by the addition of PMA/FeIII complex. The metal of the complex binds to the phosphate group, and the phenyl of the PMA group increases the excited state decay rate of the fluorescent dye. After determining the lifetimes of uncomplexed and complexed peptide (a NanoTaurus plate reader, Edinburgh Instruments, was used) conditions were tested in the presence and absence of interfering compounds. Model fitting allows determination of the percentage of phosphorylated substrate in the presence of an interfering compound which can reduce false negatives (see
figure
; fits to the average fluorescent lifetime overestimate the amount of phosphorylated peptide). High quality decay curves are required for proper model testing, which can mean increased acquisition time. However, it might be possible to re-read wells where models do not fit well using higher counts per minute. This article should provide the basis of useful software to analyze FLT data and also illustrates the modes of compound interference and their treatment in FLT measurements. Contributed by Doug Auld.
Fluorescence lifetime-based phosphorylation assays carried out in a NanoTaurus plate reader. (A) Fluorescence time responses of 9AA-labeled phosphorylated and nonphosphorylated crosstide peptide mixtures (0%–100% with 20% steps of the phosphorylated component) reacted with 2.5 mM PMA/iron(III) ligand (dots) and their fits (solid lines) by a constrained three-exponential model with fixed s1 = 1.9 ns, s2 = 5.8 ns, and s3 = 16.8 ns. Insets: Graphic representation of the si–Bi parameters. (B and D) Residual functions of all time responses shown in panels A and C, respectively. (C) Fluorescence time responses of the above peptide mixtures with the addition of an “interfering compound,” TG404 (2 lM), emitting with an 8.5-ns lifetime (dots) and their fit by a four-exponential model with fixed s1 = 1.9 ns, s2 = 5.8 ns, and s3 = 16.8 ns and free floating s4 (solid lines). (E) Assay standard curves plotted as average fluorescence lifetime versus phosphorylated fraction concentration without (solid line) and with (dashed line) fluorescence background. (F) Assay standard curves plotted as normalized B1 + B2 and B3 coefficients versus phosphorylated fraction concentration for assay samples without (circles) and with (asterisks) fluorescence background.
ITP for DNA
Kondratova VN, Botezatu IV, Shelepov VP, Lichtenstein AV. Tube gel isotachophoresis: A method for quantitative isolation of nucleic acids from diluted solutions.Anal Biochem2010; doi:10.1016/j.ab.2010.09.004.
Abstract: The technique of isotachophoresis is intended for separation of molecules having different electrophoretic mobilities in a nonhomogeneous electric field. Since the mobility of nucleic acids in water solutions is uniform and does not depend on their size (because of a uniform distribution of negatively charged phosphate groups along the molecule), isotachophoresis will concentrate rather than separate them in the mobile borderline zone between the rapid (Cl−) and the slow (β-alanine) anions. This idea served as the basis for elaboration of a novel method for isolation of nucleic acids from diluted solutions. Advantages of the method include quantitative yield (regardless of molecule size), high degree of concentration, and the ability to visually monitor the process. The method may find applications in nucleic acid isolation from highly degraded forensic and clinical samples, from bodily fluids in particular, and thereby promote development of this important direction of diagnostics.
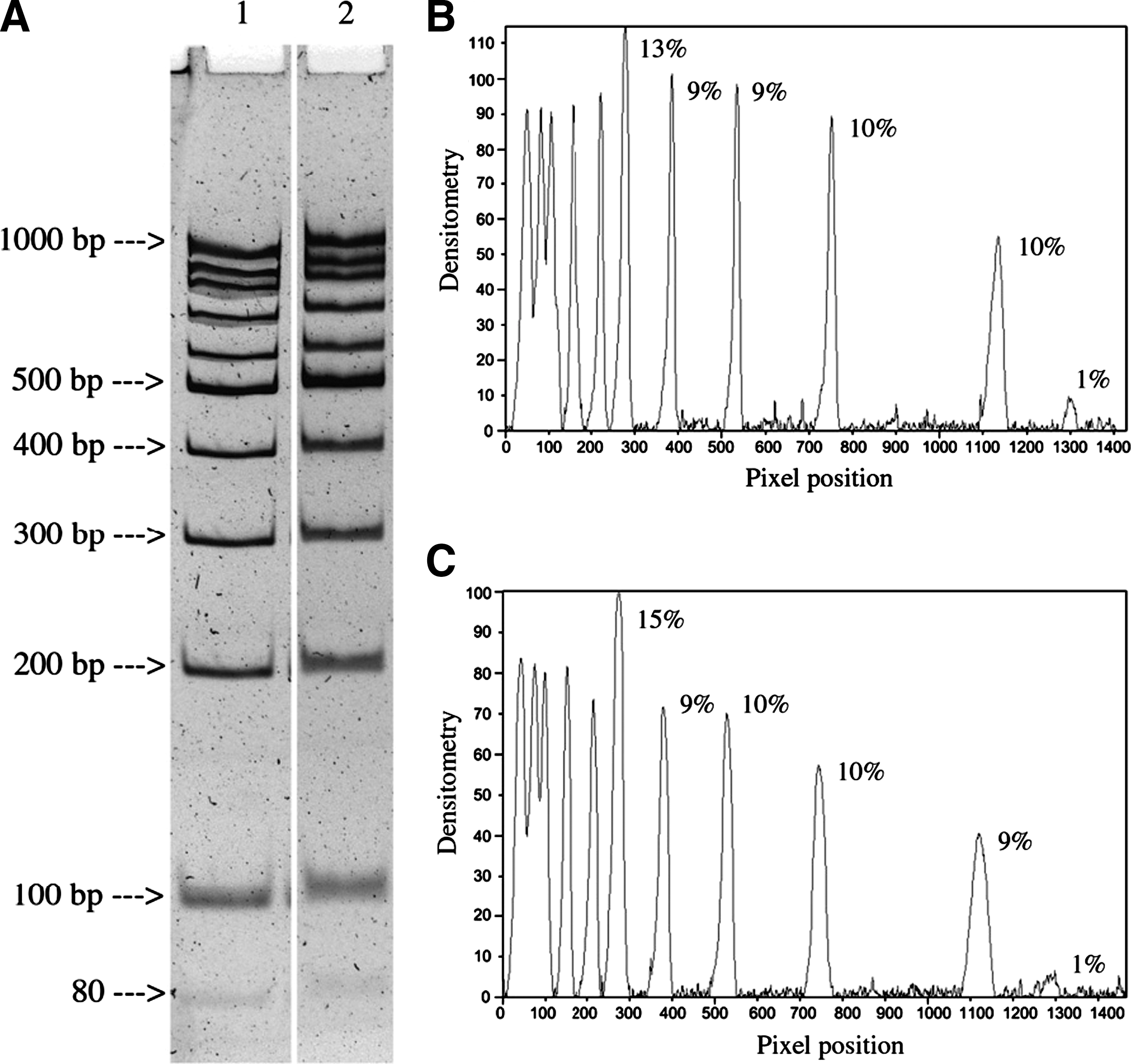
Commentary:Isotachophoresis (ITP) is a technique complementary to electrophoresis in which molecular species possessing identical electrophoretic mobility (determined by their charge-to-mass ratio) can be concentrated to an extraordinary degree due to their confinement into a narrow band created as a sharp boundary between a fast (also known as leading) electrolyte and a slow (trailing) electrolyte in the presence of an electric field. In practice, ITP is accomplished by introducing a sample (by injection or layering) between two buffers that contain the leading and trailing electrolytes as the appropriate counterions. In the present example, an ITP pair was achieved by using the rapidly migrating chloride anion in the leading buffer in combination with a slow-migrating β-alanine amino acid anion in the trailing buffer: a sample containing a negative net-charge analyte introduced between these two buffers is then subjected to an electric field with the net result being that all analyte molecules of similar electrophoretic mobility contained within the sample are further concentrated within the narrow boundary that is established between the leading and trailing buffer anions. The authors applied the ITP technique, which to date has primarily been utilized in protein analyses, to achieve a dramatic concentration of DNA contained within dilute samples derived from forensic work or in limited-cell-number diagnostic applications. A very simple device was constructed out of long-stem pipette tips (see
first figure
), which allowed the DNA concentration process to be monitored visually. With the ITP technique in its present setting, the authors demonstrated that DNA fragments of very wide range, crucially, the smallest fragments in the 50-100 bp range (see
second figure
), could be concentrated up to 200-fold at a high efficiency. In contrast, the use of traditional DNA purification kits is typically associated with significant losses of DNA when dilute samples are handled; moreover, short DNA fragments are often very poorly recovered. The present study showed that with the ITP technique not only are short DNA pieces retained at a high efficiency but the efficiency of recovery of DNA fragments of different sizes is also relatively constant (as judged by densitometry analysis of electrophoretically separated DNA after ITP concentration, second figure). This is made uniquely possible by the fact that DNA fragments, regardless of their length, carry the same charge-to-mass ratio due to the uniform distribution of phosphodiester negative charges along the nucleic acid's backbone. The present development in DNA isolation and concentration is truly exciting because it is expected to enable analyses of samples that have long been considered challenging, such as the cases of minute quantities of DNA obtained from crime scenes and clinical diagnostics being performed on a limited cell population. Contributed by Anton Simeonov.
Tube gel isotachophoresis of DNA. (A–D) One hundred nanograms of heterogeneous salmon sperm DNA (Sigma, USA) in 1 mL of 0.1× A buffer was concentrated in glass and plastic tubes. Marker dyes (xylene cyanol and bromophenol blue) were observed under visible light (images 1); DNA stained with ethidium bromide (A and B) or SYBR Gold (C–E) was observed under UV illumination (images 2). (A) Test sample in glass tube before isotachophoresis. (B) Test sample in glass tube after isotachophoresis. (C) Isotachophoresis in a plastic tube (the section with DNA band that was cut out is marked). (D) Electrophoresis in a homogeneous field (both the cathode and the anode chambers as well as 0.1% agarose gel contain A buffer). (E) Elution of DNA from a slice of 10% polyacrylamide gel (the slice was soaked for 1 h in distilled water in the presence of SYBR Gold and placed in 1 mL of 0.1× A buffer). Distribution of DNA before isotachophoresis (UV image 1) and after isotachophoresis for 3 h at room temperature (UV image 2).
Estimate of DNA recovery by tube gel isotachophoresis. (A) DNA ladder (1.5 μg in 1.5 mL of 0.1× A buffer) was isolated isotachophoretically. An aliquot (1/10 of the whole) was diluted 2.5-fold with distilled water and electrophoresed in 8% polyacrylamide gel (100 V, 4 h at room temperature) side-by-side with 150 ng of DNA ladder. Lane 1, intact DNA ladder; lane 2, DNA ladder isolated by isotachophoresis from diluted solution. (B) Densitometry of lane 1 (percentage fragment DNA is shown for the range of 80–500 bp). (C) Densitometry of lane 2 (designations are the same).
Sugar-Coating the Histone Code
Sakabe K, Wang Z, Hart GW. β-N-acetylglucosamine (O-GlcNAc) is part of the histone code.Proc Natl Acad Sci USAEarly Edition, 2010; doi:10.1073/pnas.1009023107.
Abstract: Dynamic posttranslational modification of serine and threonine residues of nucleocytoplasmic proteins by β-N-acetylglucosamine (O-GlcNAc) is a regulator of cellular processes such as transcription, signaling, and protein–protein interactions. Like phosphorylation, O-GlcNAc cycles in response to a wide variety of stimuli. Although cycling of O-GlcNAc is catalyzed by only two highly conserved enzymes, O GlcNAc transferase (OGT), which adds the sugar, and β-N-acetylglucosaminidase (O-GlcNAcase), which hydrolyzes it, the targeting of these enzymes is highly specific and is controlled by myriad interacting subunits. Here, we demonstrate by multiple specific immunological and enzymatic approaches that histones, the proteins that package DNA within the nucleus, are O-GlcNAcylated in vivo. Histones also are substrates for OGT in vitro. We identify O-GlcNAc sites on histones H2A, H2B, and H4 using mass spectrometry. Finally, we show that histone O-GlcNAcylation changes during mitosis and with heat shock. Taken together, these data show that O-GlcNAc cycles dynamically on histones and can be considered part of the histone code.
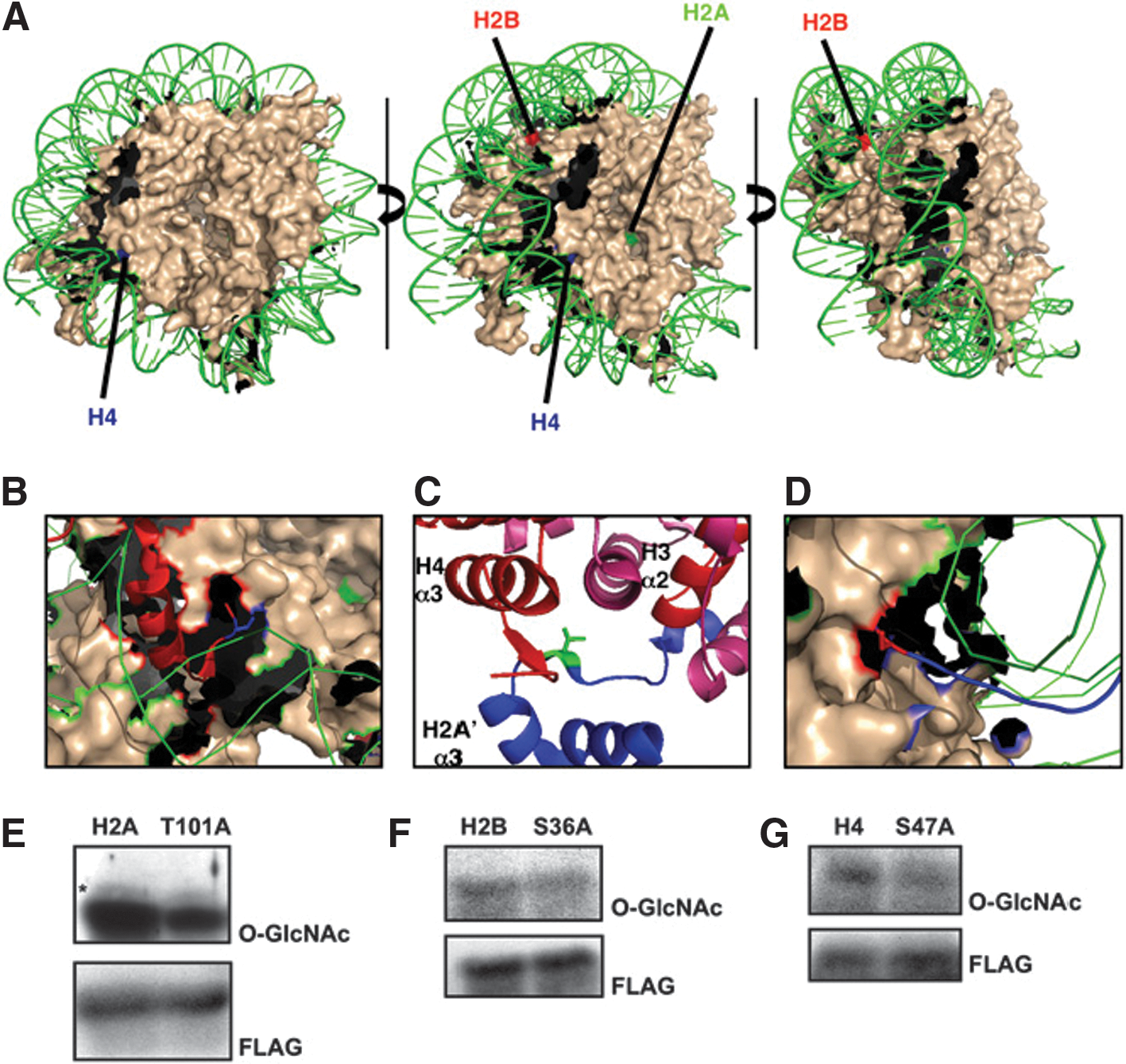
Commentary:Just when we thought that the histone code could not get any more complex, a new posttranslational modification has been added to the list. Sakabe and colleagues present multiple lines of evidence pointing to the cross-talk between protein glycosylation and transcriptional regulation. Specifically, the authors show that at least three amino acid residues on histones H2A, H2B, and H4 are dynamically modified with β-N-acetylglucosamine (O-GlcNAc), presumably by the action of O-GlcNAc transferase (OGT). Using a mutated galactosyltransferase enzyme, the authors were able to label the O-GlcNAc residues present on histones with an azide-modified galactosamine. In turn, the azide functional group installed onto O-GlcNAc was used to append a biotin tag via click chemistry. These series of modifications allowed affinity capture and quantitation of the histone proteins carrying the O-GlcNAc residues. In addition, an orthogonal detection of the O-GlcNAc group was performed by the use of anti-O-GlcNAc antibodies. Using this methodology, the authors demonstrated that human histones are indeed modified by the O-GlcNAc sugar groups (Fig. 1 in the article). Subsequently, the authors expanded their chemical strategy to map the actual sites of modification on the different histone proteins: after incorporation of the azido-sugar onto the O-GlcNAc–modified amino acids of tryptic digest–derived histone peptides followed by avidin affinity-capture and separation, the biotin disaccharide-containing peptides were subjected to a reaction sequence leading to β-elimination and dithiothreitol Michael addition, ultimately yielding DTT-labeled serine and threonine residues that could then be unambiguously identified by mass spectrometry (Supplemental Figure S2 of the article). Using this mapping strategy, O-GlcNAc modification sites within the nucleosome core particle were identified (Thr101 of histone H2A, Ser26 of histone H2B, and Ser47 of histone H4) that were in direct proximity to the interacting genomic DNA (see
first figure
) indicating that this newly identified posttranslational modification can have a direct impact on the level of compaction of the DNA around the histones and the related accessibility of genes with respect to the transcription machinery of the cell. Lastly, the authors demonstrated that overexpression of OGT, which was expected to result in increased O-GlcNAc tagging of histones, indeed caused an increased chromatin compaction (see
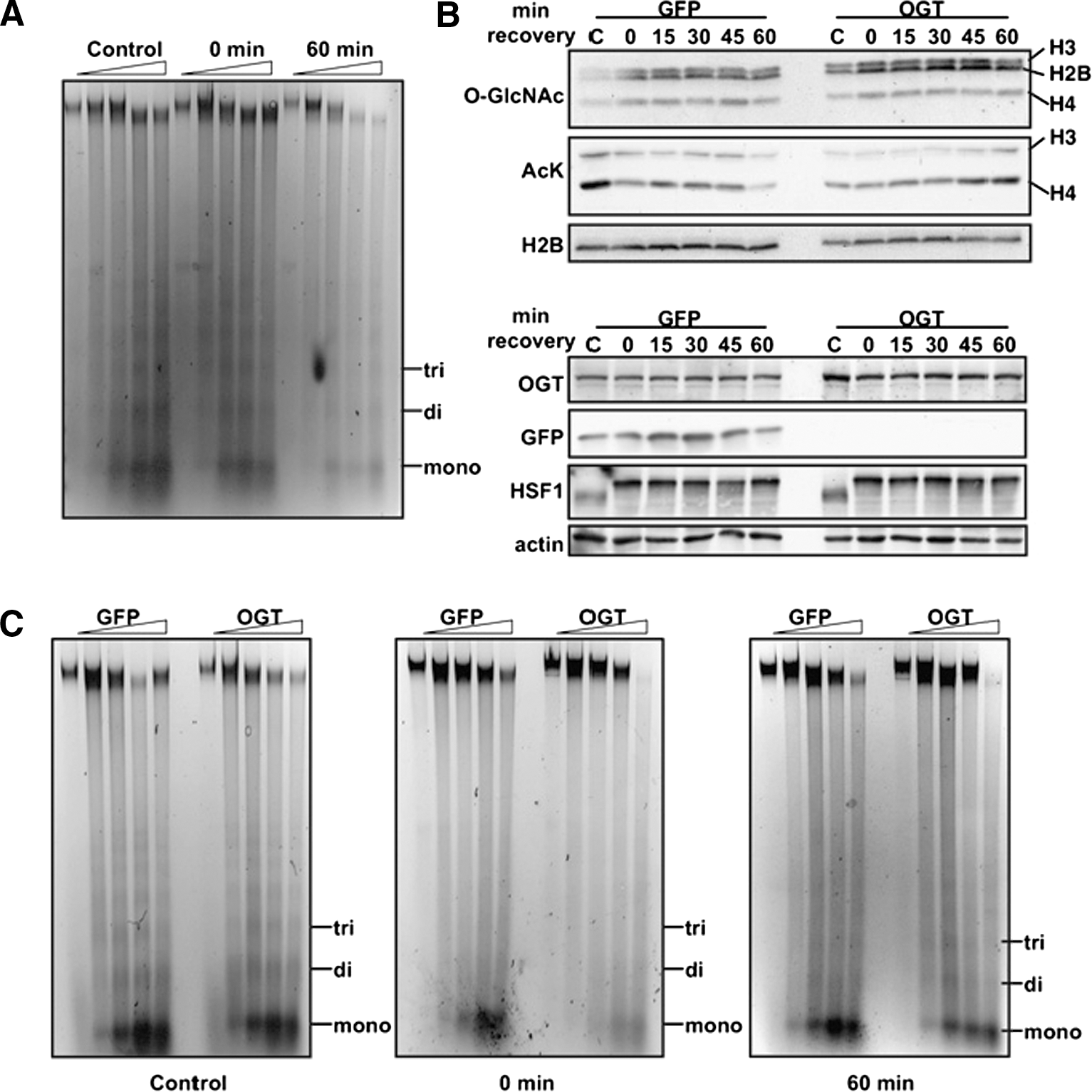
second figure
). The present studies establish O-GlcNAc modification of histones as another element of the histone code-associated transcriptional regulation and hint at the possibility of modulating posttranslational modifications for therapeutic purposes. Contributed by Anton Simeonov.
Validation of mapped O-GlcNAc sites. (A) Core nucleosomal particle is colored in nude. (For simplicity, histone tails are not shown.) DNA is in green. H4 sites are labeled in blue, H2B sites in red, and H2A sites in green. (B) Ser47 of modified H4 is shown in blue as a stick diagram. The tail of H4 is shown in red as a ribbon. (C) Thr101 of H2A is shown in green as a stick diagram, H4 is shown in red, H3 is shown in pink, and H2A is shown in blue. The α2 and α3 domains of H3 and H4 are labeled. (D) Ser36 of H2B is shown in red in a stick diagram. The histone tail of H2B is shown in blue. To verify site-mapping data, HeLa cells were transfected with FLAG-tagged constructs for wild type and corresponding Ser/Thr-to-Ala mutations for (E) H2A, (F) H2B, and (G) H4. Blots were probed for O-GlcNAc (RL2) and FLAG.
OGT overexpression affects chromatin condensation. (A) Chromatin sensitivity assays were performed. Mono-, di-, and trinucleosomes are marked. Digestions with MNase were performed for 0, 2, 5, 10, and 15 min. (B) Histones from cells overexpressing GFP or OGT exposed to 1 h heat shock at 45°C and allowed to recover at 37°C as indicated were probed for O-GlcNAc and acetylated Lys (AcK). Histones H3, H2B, and H4 are indicated. Soluble extract also was probed for OGT, GFP, HSF1, and actin. (C) Chromatin-sensitivity assays were performed on cells overexpressing GFP or OGT from control cells or cells that had been heat shocked for 1 h at 45°C and allowed to recover for 0 or 60 min at 37°C. Digestions with MNase were performed as above.
A Welcome GFP Variant
Groff D, Wang F, Jockusch S, Turro NJ, Schultz PG. A new strategy to photoactivate green fluorescent protein.Angew Chem Int Ed2010;49:7677–7679.
No abstract
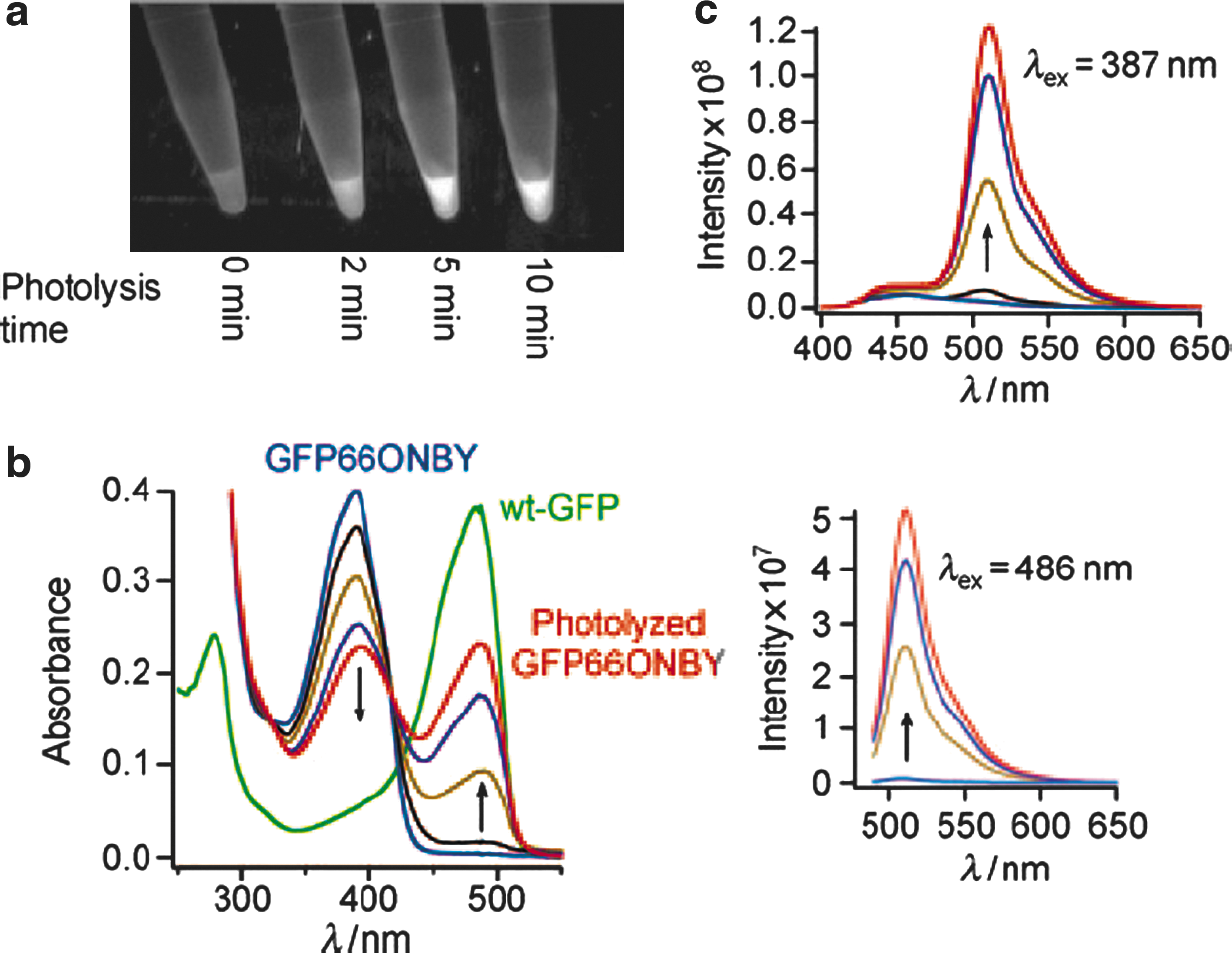
Commentary: The green fluorescent protein (GFP) from Aequorea victoria exhibits two absorbance peaks, at 395 nm (major) and 475 nm (minor), which result from the presence of neutral phenols and anionic phenolates in its chromophore, respectively. Fluorescent properties of GFP and its variants can be altered to different degrees when the molecules are subjected to intense illumination at a certain wavelength (usually at the ultraviolet or ∼400 nm spectral region) as the chromophore undergoes photoconversion and the population becomes dominated by anionic phenolates. Concomitantly, absorbance at the minor peak is enhanced and so is the emitted fluorescence. These characteristics of photoactivation underlie its utility for making molecular probes and for studying protein dynamics. PA-GFP is the first successful photoactivatable GFP variant reported that was shown to exhibit 100 times increase in fluorescence when excited at 488 nm and to remain stable for days under aerobic conditions (Patterson et al., Science, 2002;297:1873–1877). However, requirement of certain amino acid residues for photoactivation may limit the production of a variety of GFP mutants, and multiple pairs of excitation/emission wavelengths of PA-GFP can obstruct the variant's use for other applications. Herein, the study by Schultz's lab provides a novel and welcome GFP variant, termed GFP66ONBY. The authors took advantage of a nonnatural analogue of tyrosine, o-nitrobenzyl-O-tyrosine (ONBY), to replace tyrosine 66 in the chromophore by site-directed mutagenesis. The initial low fluorescence in GFP66ONBY was speculated to be due to photo-induced electron transfer (PET), and restoration of fluorescence was obtained by irradiation at 365 nm to remove the o-nitrobenzyl group (Scheme 1 in the article). To illustrate their strategy, purified GFP66ONBY was irradiated with 365-nm light for different time periods, and a bathochromic shift was observed at 486 nm with its peak height proportional to the time of irradiation treatment (see
first figure
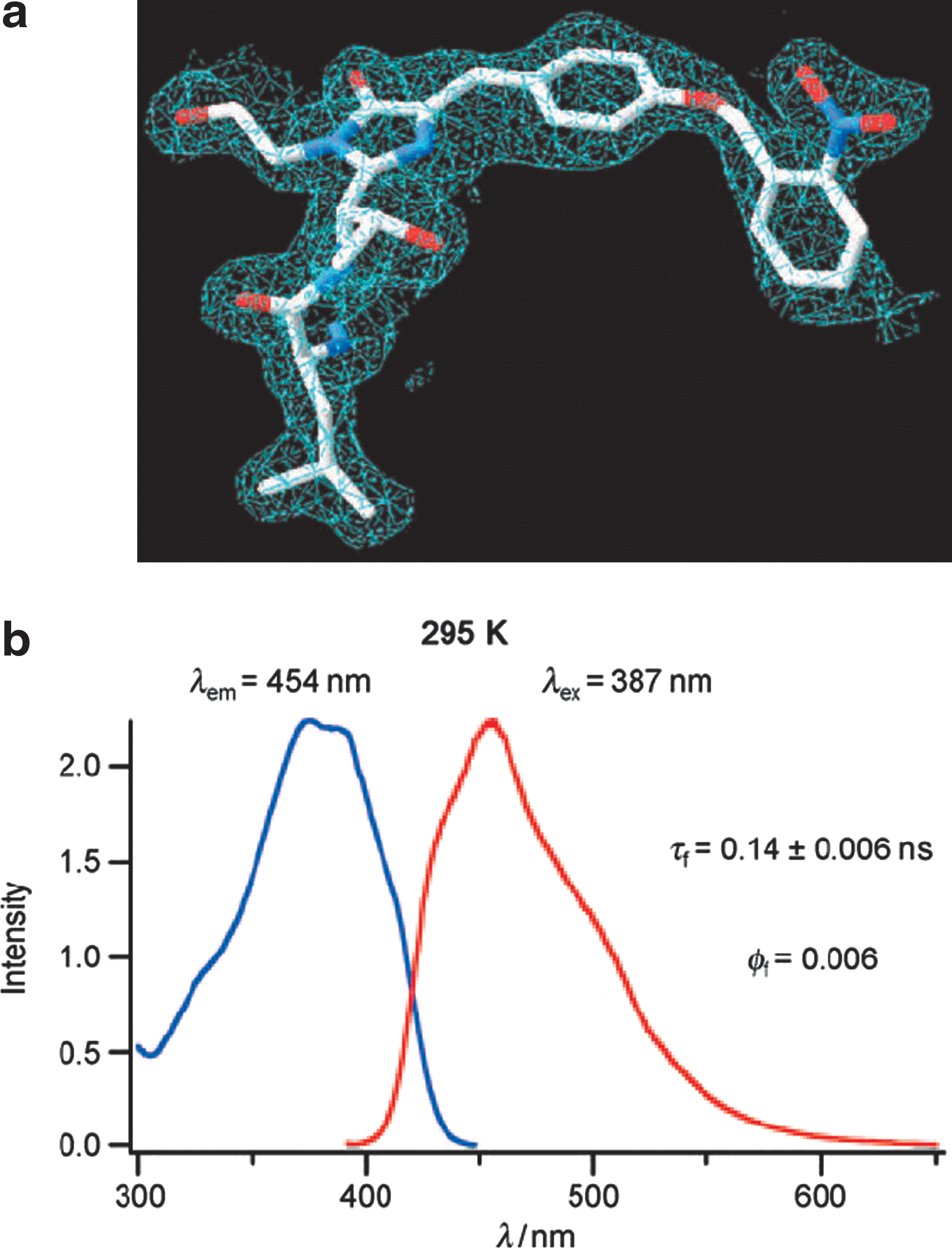
). In addition, the extinction coefficient and relative brightness of GFP66ONBY were estimated to have increased at least 100 times and four orders of magnitude upon photolysis, respectively. Other desirable properties of GFP66ONBY include its photostability and relatively long fluorescent lifetime (2.4 ns). To further support that the dark form of the GFP66ONBY (see
second figure
, panel b) was most likely due to PET instead of other factors, such as the absence of a matured fluorophore, crystal structure was subsequently obtained (see
second figure
, panel a). It was shown from the crystal structure that both the o-nitrobenzyl group and the benzylidene imidazolinone, components necessary for backbone cyclization and fluorophore maturation, were clearly present. Overall, this proof-of-concept study provides a new and improved version of a GFP variant that is promising in protein and imaging applications. Dependence on any specific residue in the chromophore is not needed for photoactivation, and this variant may also enable multiplexing technologies with other GFP variants (see ADDT commentary “Managing Oligomerization for Fluorescent Protein Assays”2007;5(4):478). Recently, the incorporation of unnatural amino acid in proteins has been shown to be feasible in mammalian cells (Thibodeaux et al., PLoS ONE, 2010;5 (6) e11263). Thus, studies utilizing GFP66ONBY in both prokaryotic and eukaryotic cells are expected to follow in the near future. Contributed by Wendy Lea.
GFP66ONBY is nonfluorescent until irradiation with 365 nm light. (a) GFP66ONBY purified using Ni–NTA was irradiated using a Spectra Physics 500 Watt lamp with a 365 nm filter for the indicated lengths of time and then visualized using a UV transilluminator. (b) UV/Vis absorbance spectra of GFP66ONBY upon photolysis. (c) Emission spectra of photolyzed GFP66ONBY excited at 387 nm (top) and 486 nm (bottom). For (b) and (c), samples were photolyzed at 365 nm for 3 (black), 23 (brown), 43 (purple), and 60 min (red) using a 4-W UVP lamp.
GFP66ONBY fluorescence is likely quenched through photon induced electron transfer to the o-nitrobenzyl group. (a) His-tagged GFP66ONBY was purified with affinity chromatography and crystallized to yield crystals that diffracted to 2.1 Å. Electron density from the X-ray structure clearly shows the presence of the o-nitrobenzyl group and cyclized backbone. (b) Fluorescence excitation (blue) and emission (red) spectra of caged GFP66ONBY in Tris buffer (pH 8) at room temperature; caged GFP66ONBY is practically nonfluorescent (fluorescence quantum yield = 0.006) and shows only a short fluorescence lifetime (0.14 ± 0.06 ns).
Dendritic Drug
Dernedde J, Rausch A, Weinhart M, Enders S, Tauber R, Licha K, Schirner M, Zügel U, von Bonin A, Haag R. Dendritic polyglycerol sulfates as multivalent inhibitors of inflammation.Proc Natl Acad Sci USA2010; doi: 10.1073/pnas.1003103107.
Abstract: Adhesive interactions of leukocytes and endothelial cells initiate leukocyte migration to inflamed tissue and are important for immune surveillance. Acute and chronic inflammatory diseases show a dysregulated immune response and result in a massive efflux of leukocytes that contributes to further tissue damage. Therefore, targeting leukocyte trafficking may provide a potent form of anti-inflammatory therapy. Leukocyte migration is initiated by interactions of the cell adhesion molecules E-, L-, and P-selectin and their corresponding carbohydrate ligands. Compounds that efficiently address these interactions are therefore of high therapeutic interest. Based on this rationale we investigated synthetic dendritic polyglycerol sulfates (dPGS) as macromolecular inhibitors that operate via a multivalent binding mechanism mimicking naturally occurring ligands. dPGS inhibited both leukocytic L-selectin and endothelial P-selectin with high efficacy. Size and degree of sulfation of the polymer core determined selectin binding affinity. Administration of dPGS in a contact dermatitis mouse model dampened leukocyte extravasation as effectively as glucocorticoids did and edema formation was significantly reduced. In addition, dPGS interacted with the complement factors C3 and C5 as was shown in vitro and reduced C5a levels in a mouse model of complement activation. Thus, dPGS represent an innovative class of a fully synthetic polymer therapeutics that may be used for the treatment of inflammatory diseases.
Commentary:A number of inflammatory diseases are associated with excessive recruitment of leukocytes to inflamed tissue. The binding of leukocytes to activated endothelium is achieved via multiple extended-surface type interactions between selectins and carbohydrate residues on the surface of glycoproteins present on leukocytes and the endothelial cells. Disruption of such interactions is expected to yield a beneficial therapeutic effect by decreasing the extent of leukocyte recruitment and subsequently slowing tissue destruction in autoimmune disorders. In the present work, dendritic polyglycerols (dPG) decorated with multiple sulfate groups were developed and tested as inhibitors of leukocyte recruitment to sites of inflammation. The novel macromolecules were synthetically derived and shown to be biocompatible; due to their tree-like (dendritic) structure (see
first figure
), multiple functional groups could be attached to their termini. In the present application, sulfate groups were appended to variable-core dPGs and the binding of the resulting agents to P- and L-selectin proteins, known to possess extended positively charged surfaces, was evaluated by surface plasmon resonance. As anticipated, nanomolar-level affinities were obtained from the optimized macromolecules (see
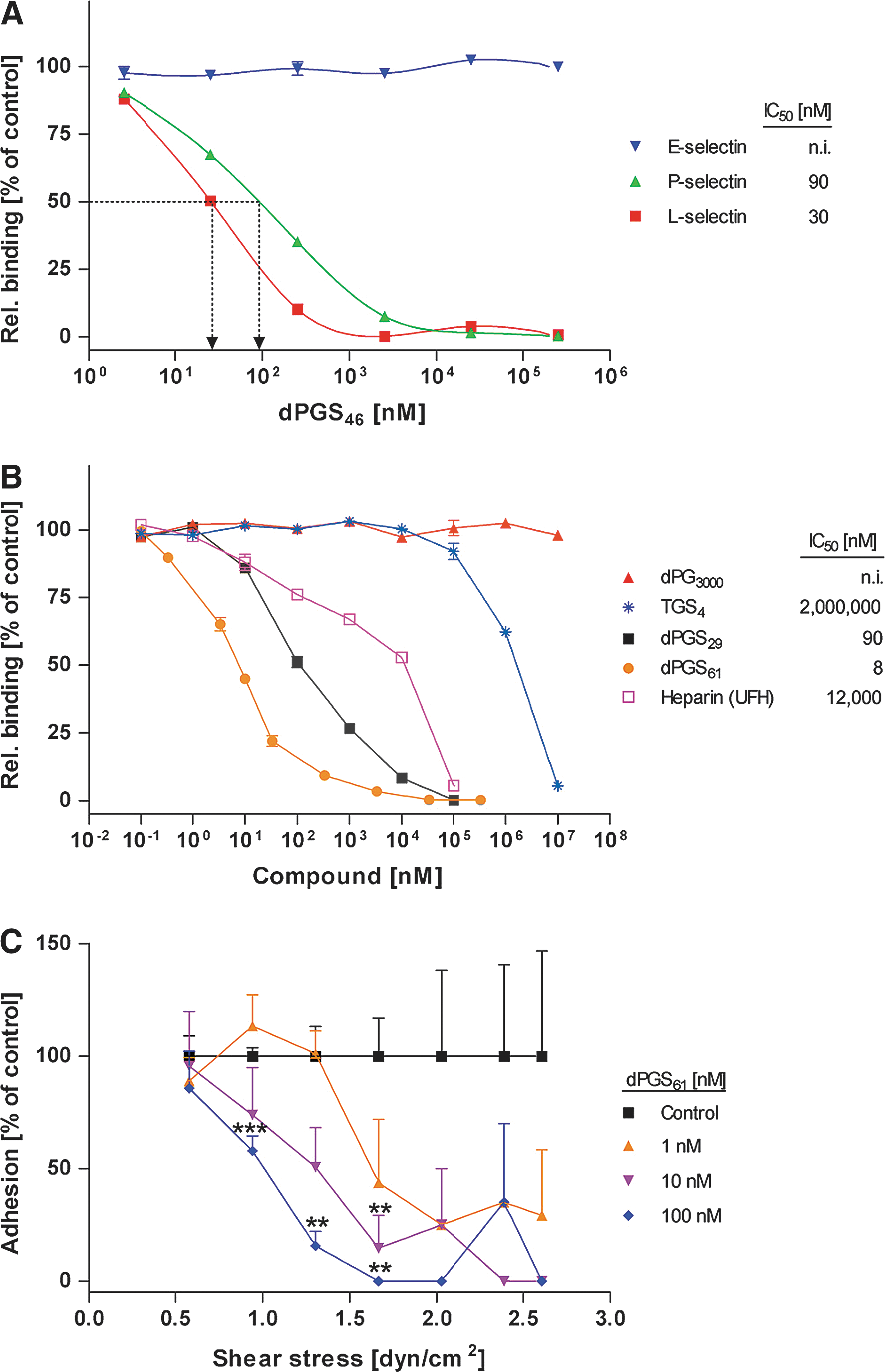
second figure
). Furthermore, the macromolecule dPGS61 was shown to efficiently inhibit binding of L-selectin–expressing cells to TNF-α–activated human umbilical vein endothelial cells in a flow chamber model of human blood flow (see
second figure
). Lastly, the effect of sulfated dPG in an in vivo skin inflammation model was tested with a mouse acute allergic contact dermatitis induced by an application of the allergen trimellitic anhydride, with the agent's effect being determined by measuring the neutrophil elastase activity in the mouse ear homogenates (Fig. 3 of the article): a significant reduction in neutrophil elastase activity was detected at dPGS61 dose as low as 3 mg/kg. The success of these proof-of-concept studies of dPGS establishes these unique dendritic polymers as a promising new class of therapeutic agents to target biological processes mediated by extended-surface protein–sugar interactions. Contributed by Anton Simeonov.
Dendritic polyglycerol sulfate structure. Representative and idealized structure of dPGS. Sulfate end groups are shown in red. All details of the different dPGS molecules are summarized in Table S1 of the article.
In vitro selectin binding studies of dPGS with different degrees of sulfation. (A) dPGS, here dPGS46, act as competitive selectin binder and addresses L- and P-selectin, but not E-selectin. (B) The number of sulfate groups per molecule and the core size determine selectin inhibition efficacy (shown for L-selectin). (C) Dose-dependent inhibition of leukocyte binding to HUVECs in the flow chamber. Depending on the shear stress and the amount of dPGS applied, leukocyte adhesion decreased. L-selectin transfected NALM-6 cells were preincubated with dPGS61 (0, 1, 10, and 100 nM) and perfused into the system. Adhesion to activated HUVECs was determined by cell counting. As a control, the number of untreated adherent cells was set to 100%. The flow chamber geometry allowed to measure adhesion at defined shear stresses between 0.6 and 2.7 dyn/cm2. Mean values ± SEM, n = 4. Student's t-test was used for statistical comparison. ***P < 0.001, **P < 0.01, *P < 0.05.