
Abstract
Histone methylation is a regulated feature of nucleosomes that can have an impact on gene expression. The methylation state of histone residues has also been found in recent years to be associated with various disorders. Tools for detecting methylation state changes are very useful for dissecting the function of these epigenetic marks. In this work, a sensitive homogeneous assay for histone demethylase activity at the H3K4 site has been developed in a time-resolved fluorescent resonance energy transfer assay format. The assay is based on the detection of the unmethylated H3 peptide by a fluorescent europium-chelate labeled monoclonal antibody binding specifically to the H3K4 site. The assay was validated for histone lysine-specific demethylase 1 and was demonstrated to be a suitable assay for inhibitor profiling and high-throughput screening.
Introduction
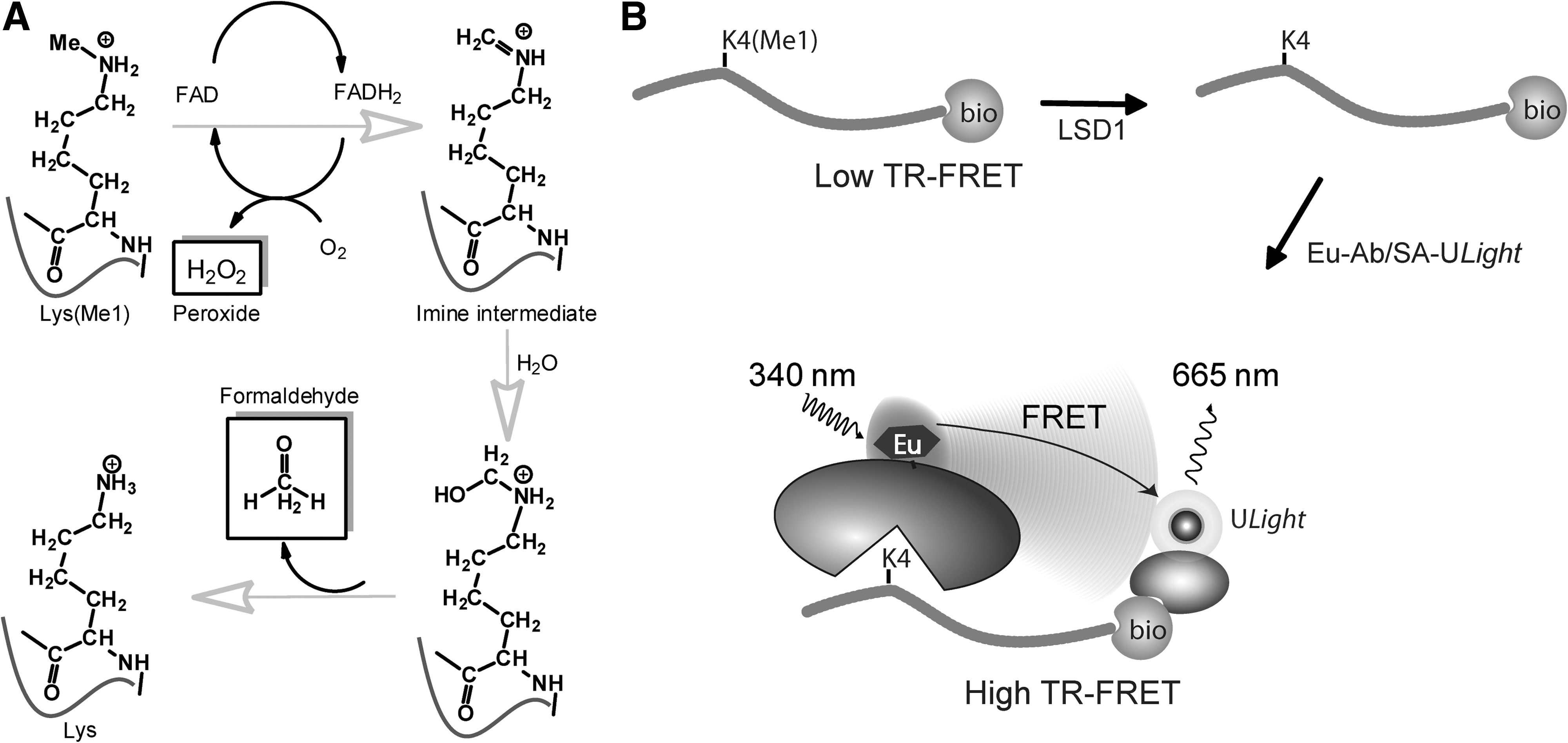
The first lysine-specific histone demethylase discovered was LSD1 (also known as KDM1, AOF2). 11 Discovery of other KDMs soon followed. These KDMs are divided into two families with different catalytic mechanisms: one family represents flavin-dependent amine oxidases; the other contains the Jumonji C domain. LSD1 is a member of the former monoamine oxygenase family that catalyzes the demethylation of mono- and di-methylated H3K4 through the formation of a positively charged imine intermediate (Fig. 1A). The unstable imine degrades to produce formaldehyde and the amine. In the reaction, flavin adenine dinucleotide (FAD) is reduced to FADH2, which is subsequently reoxidized by molecular oxygen with the production of hydrogen peroxide (Fig. 1A). 13
Several biological assay systems have been used for measuring the enzymatic activity of KDMs, from direct measurement of modified histone product by immuno-detection (western blot, enzyme-linked immunosorbent assay [ELISA]) and liquid chromatography/mass spectroscopy (LC-MS) detection 14 to coupled enzymatic reactions that detect the by-products hydrogen peroxide or formaldehyde. 13 Each of these approaches has some advantages and limitations. For example, using the LC/MS method for direct detection of methylation or demethylation allows the use of histone tail peptide, histone protein, or nucleosomes as substrate. In the case of histones or nucleosome substrates, proteolytic digestion may be required before LC/MS analysis. Additionally, the detection sensitivity with LC/MS may require the use of higher substrate and enzyme concentrations, and the throughput of most LC/MS assays is limited although a recent report demonstrated an LC/MS-based screening of a metabolic enzyme against a 800,000 low-molecular weight (LMW) library. 15 At present, most of the commercial assay kits for demethylases are based on coupled enzymatic assays that measure either formaldehyde or hydrogen peroxide formation. Although these coupled assays are generic and work well under certain conditions, the enzyme and substrate concentrations used in the assays suggest that the detection sensitivity of these assays may be more limited than direct detection of methylation states. In addition, rigorous counterscreens have to be used to eliminate compounds that simply interfere with the coupled assay system. Radioactive assays such as the use of [ 3 H]-methyl labeled histone substrates have been reported with detection by scintillation counting. 16 This method is quite sensitive but has all the drawbacks inherent in radioactive assays. There are also fluorescent assays reported based on microfluidic detection. 17 Due to throughput and/or sensitivity limitations of these assays, there is a strong need for the development of more sensitive, simple, and high-throughput assays for KDMs.
The time-resolved fluorescent resonance energy transfer (TR-FRET) assay is one of the most common and preferred assay methods in high-throughput screening (HTS) due to its homogeneous and high-throughput format, good sensitivity, and robust time-delayed signal detection. 18,19 For KDMs, the assays currently available are based on antibodies that recognize methylated peptides and, therefore, measure either a demethylated intermediate or the methylated substrate in a signal decrease mode of detection. However, a more desirable assay format is one where demethylases activity is measured in a signal increase mode of detection. One of the limitations for implementing such an assay for KDMs has been the availability of suitable site-specific and methylation state-specific antibodies. Recently, the development of monoclonal antibodies that are specific in recognizing unmethylated lysines in a site-specific manner have been reported for histones. 20,21 This opens the possibility of developing a TR-FRET type of homogeneous assay for demethylases with signal increase readout for formation of the demethylated peptide product. In this article, we describe a collaborative effort to develop such an assay by using a sensitive and homogeneous TR-FRET assay format. For detecting demethylation at the histone H3K4 site, we developed a europium (Eu)-labeled monoclonal antibody that specifically binds to the unmethylated H3K4 site. We tested this approach with a TR-FRET assay for LSD1 and demonstrate that this assay is sensitive and suitable for use in HTS and compound characterization (Fig. 1).
Materials and Methods
Materials
The N-terminal glutathione-S-transferase-tagged histone demethylase LSD1 (aa158-852) was obtained from BPS Bioscience (San Diego, CA. Cat. no. 50100). The monomethylated histone substrate peptide used in this article was a synthetic peptide corresponding to amino acids 1–21 of human histone H3 with three amino acids and a biotin motif (GGK-biotin) added to the C-terminus (sequence: ART-K(Me1)-QTARKSTGGKAPRKQLA-GGK-biotin), with 95% purity according to the manufacturer specification (GL Biochem Ltd., Shanghai, China; Cat no. 217548 or from AnaSpec Cat. no. 61702). Other H3K4Me1-3, H3K9Me1-3, and H3R2Me1 peptides for Eu-labeled antibody specificity experiments were from AnaSpec (Cat. no. 64355-025, 64356-025, 64357-025, 64358-025, 64359, 64360, 64629, respectively), and three additional H3 (aa 21–44) peptides (H3K27Me0/1, Cat. no. P100128-SY183291 and P100202-ML183289, respectively; H3K27Me2, Millipore, Cat. no. 12-566). The reference inhibitor peptides H3M4 (K→M mutation) and H3R4 (K→R mutation) were also from GL Biochem Ltd. (Cat. no. 184422 and Cat no. 217553, respectively). The reference inhibitor compound tranylcypromine (or trans-2-phenylcyclopropylamine hydrochloride, also known as, 2-PCPA) was from Sigma (St. Louis, MO. Cat. no. P8511). The TR-FRET acceptor reagent ULight ™-labeled streptavidin (ULight-SA,Cat. no. TRF0102) and detection buffer (lanthanide chelate excite [LANCE®] detection buffer, 10×, Cat. no. CR97-100) were from PerkinElmer (Montreal, Quebec, Canada). The experiments that were carried out in a 384-well plate used white, low-volume 384-well plates (ProxiPlate™-384 Plus from PerkinElmer, Cat. no. 6008280). The coupled LSD1 assay reagent horseradish peroxidase (HRP) was from Cayman Chemical Co. (Ann Arbor, MI). Experiments carried out in a 1,536-well plate format used a black Greiner plate (Cat. no. 782076). All other chemicals and reagents were purchased from various manufacturers at analytical or higher grades.
Eu-Labeled Anti-H3K4Me0 (Unmethylated) Monoclonal Antibody Preparation
A Eu-labeled mouse monoclonal antibody (Eu-antibody) specific to unmodified Histone H3K4 (H3K4Me0; PerkinElmer Cat. no. TRF0404) was used in TR-FRET assays. The antibody was labeled with W1024 Eu-ITC chelate (PerkinElmer) in carbonate buffer (pH 9.3) for 16 h at 4°C, and purified over a G-50 column. The number of Eu-chelate molecules per antibody was spectrophotometrically estimated by considering the molar extinction coefficient of both the antibody at 280 nm and Eu-chelate at 335 nm. The labeled antibody was resuspended at a concentration of 0.625 μM in 50 mM Tris-HCl (pH 7.4), 0.9% NaCl, 0.1% bovine serum albumin (BSA), and 0.05% sodium azide as preservative. Antibody lots with Eu-labeling ratios between 6.5 and 8.0 were used throughout this study.
TR-FRET Assay Development
Peptide specificity/selectivity of the Eu-labeled anti-H3K4Me0 antibody (Eu-antibody) was tested. The labeled antibody was tested for detection of 3 methylated H3 peptides at K4 position and H3 methylated at K9, R2, and K27 positions (see Materials and Methods section), together with the unmethylated H3 peptides at varying concentrations (from 10−5 to 10−10 M). The experiment was tested in triplicate in a white OptiPlate 384-well plate, with 10 μL of biotin peptides diluted in 50 mM Tris-HCl (pH 8.0) buffer mixing with 10 μL of a mix of Eu-Ab 2×(final 2 nM) and ULight-SA 2×(final 50 nM), both diluted in 1×LANCE detection buffer. The mixture was allowed for incubation for 60 min at room temperature (RT) and read with an EnVision® Multilabel Reader (PerkinElmer) by using a time-resolved fluorescence detection protocol (Ex=340 nm and Em=615 and 665 nm) without the top seal.
The initial assay conditions were tested with the unmethylated peptide (H3K4Me0) at 10 nM concentration by using varying amounts of Eu-antibody (0.25, 0.5, 1, and 2 nM final concentration) and ULight-SA (10, 20, and 50 nM) in the 1×LANCE detection buffer. The assay was conducted in a low-volume 384-well plate by mixing 10 μL of peptide and 10 μL of detection mixture. The plate was read on EnVision by using the same detection protocol as just indicated after incubation at RT for 60 min. One nM Eu-antibody and 10 nM of ULight-SA were selected as detection conditions for the following experiments.
Assay buffer was further optimized by a design of experiment (DOE) protocol, including testing of several salt and pH conditions, as well as several buffers and additives (i.e., reducing reagents, BSA, detergents, etc.). The optimized assay buffer for assay development experiments contained 20 mM Tris-HCl, pH 8.0, 2% glycerol, 0.1% BSA, 0.5 μM FAD, 0.5 mM tris(2-carboxyethyl)phosphine, and 0.02% Tween-20. Using this optimized buffer system, an LSD1 titration and a reaction time course were conducted in the same experiment with the LSD1 amount varying from 2 to 0.031 nM in a 2-fold dilution series, and the reaction time varied from 15 to 60 min at 15 min intervals. Reactions were started by adding 5 μL of enzyme to 5 μL of peptide substrate. The reactions were stopped by addition of detection reagent including 100 μM of tranylcypromine in the detection mixture. The reactions were detected and read as just stated.
Dimethyl sulfoxide (DMSO) tolerance of the assay was determined by testing a range of DMSO concentrations from 0% to 5% in the reaction mixture. First, substrate peptide was diluted in DMSO containing assay buffer, then 5 μL of enzyme was added to 5 μL of peptide containing various amount of DMSO. End-point detection after 30 min reaction was used in the test.
For known reference inhibitor concentration-response curve titrations, compounds were preincubated with enzyme for 30 min before adding peptide substrate. Tranylcypromine was titrated from 200 μM in a 2-fold dilution series, whereas inhibitor peptides H3M4 and H3R4 were titrated from 1 μM. The reaction was detected and read as just stated.
Coupled LSD1 Assay
LSD1 was titrated from 1,000 nM (2-fold dilutions, also including 0 nM as a control) in a coupled LSD1 fluorescence assay. The coupled assay utilizes the hydrogen peroxide (H2O2) by-product, and it was detected by the generic Amplex Red detection coupled with HRP. The protocol used is the following. The 20 μL reaction included 0.4 μM HRP, 100 μM Amplex Red, and LSD1 enzyme. Fifty micrometer of peptide H3K4Me1 (or assay buffer for the control experiment) was added to start the reaction. The plate was incubated for 60 min at RT and read with Ex=535 nm and Em=590 nm. Two sets of data were generated with or without peptide substrate present in the reaction.
Data Analysis
Curve fitting for enzyme kinetics and inhibitor dose response were performed by using the commercial software GraphPad Prism. Correlation scatter plots were generated in Spotfire data visualization software. Percent inhibition of sample was calculated and normalized by the uninhibited control (high control) and fully inhibited control (low control) reactions with the formula Inhibition %=(high control−sample)/(high control−low control)×100.
Results
Assay Configuration, Set Up, and Substrate Specificity
The TR-FRET assay configuration is illustrated in Figure 1B. Monomethylated H3K4 peptide (H3K4Me1) was used as the substrate, which is tagged with biotin at the C-terminus. On demethylation at K4 by KDM (e.g., LSD1 in this case), the reaction generates unmethylated H3K4 peptide. We have used a mouse monoclonal anti-unmethylated H3K4 site-specific antibody covalently labeled with Eu chelate (Cat. No. TRF0404, PerkinElmer) to specifically detect the unmethylated H3K4 product. The TR-FRET signal is generated by the binding of this Eu-labeled antibody as the donor and ULight-SA as the acceptor to the same peptide product.
To confirm the labeled antibody specificity toward different methylation states at the K4 lysine site and its selectivity toward different lysine methylation sites on the histone H3 peptide, we first tested six H3 peptides with 1 to 3 methylation states at K4 and K9 positions, and an H3R2Me1 peptide, together with the unmethylated H3 peptides, in the TR-FRET assay format. The results are shown in Figure 2. For a broad range of peptide concentrations tested in this study (see Materials and Methods section), TR-FRET signal was generated only with peptides unmethylated at K4 position. Mono-, di-, and tri- methylated peptides at K4 do not show any significant signal at all tested peptide concentrations. This result indicates specific methylation of K4 (Me1/2/3) blocks binding of the Eu-antibody to the unmodified K4 residue, whereas other modifications (i.e., methylations) tested (H3K9Me1/2/3, H3R2me1) have no effect on Eu-Ab binding. In addition, three biotinylated H3 peptides lacking the K4 position (H3K27Me0/1/2, aa21–44) were also tested, and all of these did not show any binding signals (data not shown). This result is consistent with the previous tests which show that such an antibody is only sensitive to modifications at K4 site (and possibly T3 site, see Discussion section) of the H3 peptide. 20

Specificity and selectivity of the Eu-labeled anti-H3K4Me0 antibody against methylated H3 peptides as measured in TR-FRET assay format.
From the peptide titration test, it was determined that the assay signal is linear for the unmethylated K4 peptide up to about 60–100 nM with the amount of detection reagents (concentration) used in the experiment. We have used 60–100 nM monomethylated H3K4 peptide as substrate in our subsequent LSD1 assay development to ensure that the assay signal generated is linear for the peptide product formed. In addition, this substrate level is well below the KM values reported for the H3K4me1–2 peptide for LSD1, 17,22 –24 to further ensure the sensitivity of the assay for inhibitor test and HTS. The product conversion at these assay conditions was estimated to be <30%. The assay was also optimized for the usage of detection reagents (Eu-antibody and ULight-SA) for maximized assay signal with minimized reagent consumption. From the results, 1 nM Eu-antibody and 10 nM ULight-SA were selected for donor and acceptor reagents, respectively.
TR-FRET Assay Development for LSD1
To demonstrate the suitability and characteristics of the TR-FRET assay for use in LSD1 inhibitor measurement and HTS, we utilized a commercially available LSD1 as a test case. A time course (Fig. 3A) and an enzyme titration (Fig. 3B) for the LSD1 TR-FRET assay was carried out in a low-volume 384 multi-well plate format (experimental procedures in Table 1). The assay buffer used in the experiments was optimized from a test run employing a DOE procedure. From the titration results, the linear signal ranges for LSD1 and reaction time were obtained. For instance, at 0.5 nM LSD1, the assay signal is linear up to about 15 min. At 0.25 nM and 0.125 nM LSD1, the signal linear ranges are up to 30 and 60 min, respectively. We chose the 0.25 nM LSD1 and a reaction time of 30 min for most of the test runs. For high-throughput inhibitor testing, we chose to use 0.125 nM enzyme and 60 min reaction in an end-point readout assay format for ease of implementation of the high-throughput assay (Fig. 3).
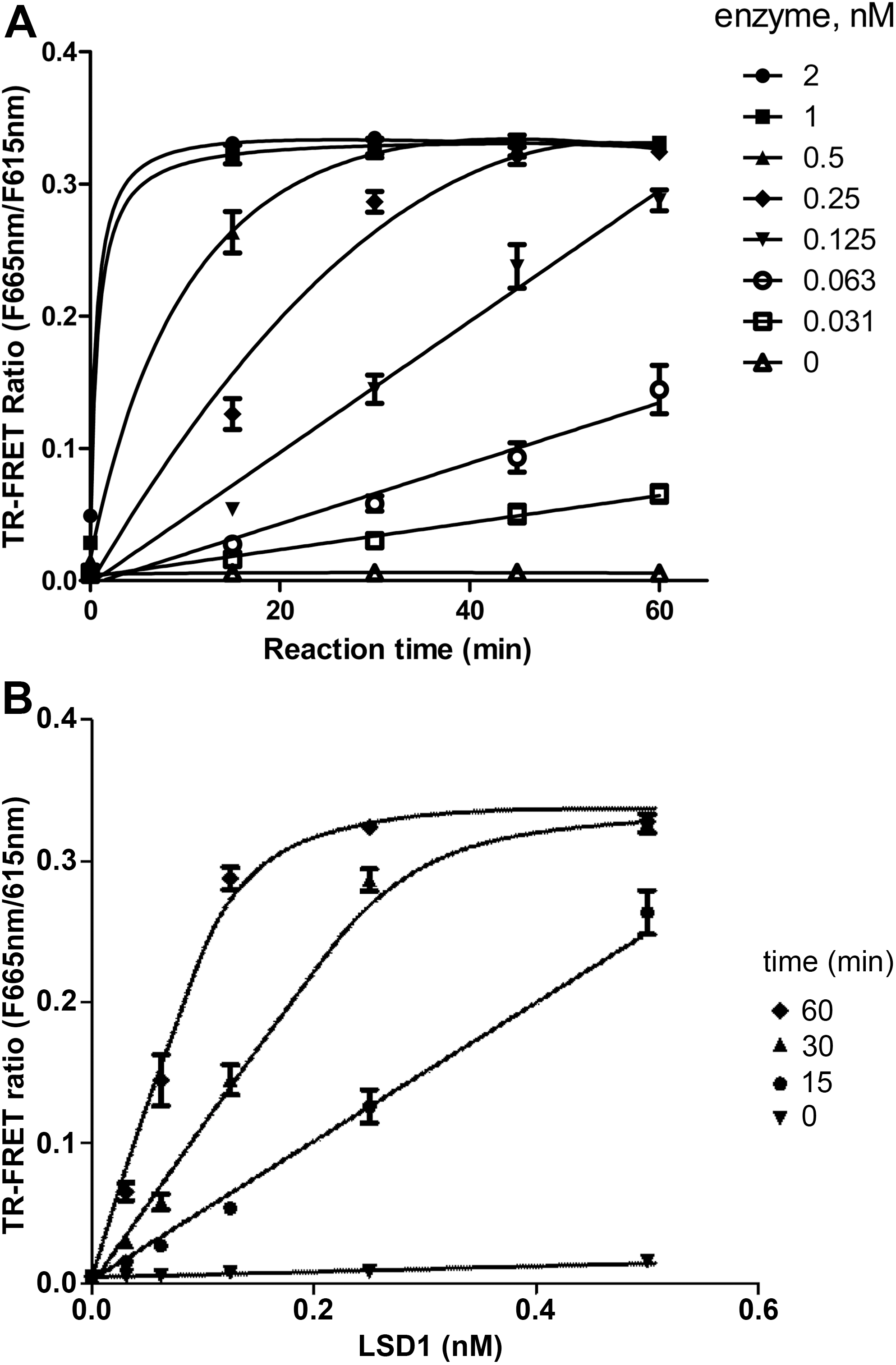
Enzyme titration and time course for the LSD1 TR-FRET assay (in triplicate, mean±SD).
384w- (1,536w-) Plate Assay Protocols
Step Notes
Volumes are for low-volume 384w plates or in parentheses 1,536w-plates.
1. Assay buffer: 20 mM Tris-HCl, pH 8.0, 2% glycerol, 0.1% BSA, 0.5 μM FAD, 0.5 mM TCEP, and 0.02% Tween-20. Final peptide concentration is 60 nM.
2. Via acoustic addition, pre-plated in the assay plate.
3. Final concentration of LSD1 is 0.5 nM.
4. Plate is sealed (optional) during incubation time.
5. In 1×LANCE® detection buffer.
6. Plate is sealed (optional) during incubation time.
7. Perkin Elmer EnVision® Multilabel Reader; Ex=340 nm and Em=615 and 665 nm.
2-PCPA, trans-2-phenylcyclopropylamine; BSA, bovine serum albumin; DMSO, dimethyl sulfoxide; FAD, flavin adenine dinucleotide; LSD1, lysine specific demethylase 1; TCEP, tris(2-carboxyethyl)phosphine; TR-FRET, time-resolved fluorescent resonance energy transfer.
Before performing small-molecule inhibitor testing and screening, the tolerance to DMSO solvent was determined for the assay. A DMSO titration experiment indicates that DMSO levels from 0% to 2.5% have an insignificant effect on the signal of the TR-FRET assay for LSD1 under the testing conditions (data not shown). This implies that the assay signal is robust toward routine testing DMSO levels that are usually maintained at a level of <2% in the assay.
To further validate the assay performance and sensitivity, we tested the inhibition profile of several known LSD1 inhibitors. Both covalent inhibitors such as tranylcypromine (2-PCPA) and noncovalent inhibitors, that is, peptide substrate analogs, were included in the tests. The inhibitor titration results are shown in Figure 4. The IC50 value for tranylcypromine is estimated to be approximately 10–20 μM, in the close vicinity of a recently reported value (32 uM). 25 It should be noted, however, with such covalent inhibitors that the inhibition potency is time dependent for the first hour or so of preincubation of the inhibitor and the LSD1 enzyme (data not shown). Both H3M4 (K to M mutation of the H3 peptide) and H3R4 (K to R mutation of the H3 peptide) showed potent submicromolar inhibition, with IC50 values determined to be 18 and 55 nM in the TR-FRET assay, respectively. Both the potency and rank order of the two inhibitor peptides are in good agreement with the previously published inhibition profile. 26 Both inhibitor peptides show either no or minimal binding (signal interference) up to 20 μM in the TR-FRET assay. The assay quality based on the noninhibited and fully inhibited testing points (which serve as high and low controls, respectively) is high, with the assay data quality statistical parameter Z′ factor 27 of 0.8 or higher from the tests. These results strongly suggest that the TR-FRET assay developed here is sensitive to both covalent and noncovalent inhibitors and is suitable for measuring inhibitor potency profile. We have compared the current LSD1 TR-FRET assay with a coupled LSD1 assay based on H2O2 detection. Based on the required amounts of enzyme (0.1–1 nM in TR-FRET, Fig. 3B vs. 10–100 nM in H2O2 detection, Fig. 5) and substrate (50–100 nM in TR-FRET vs. 20–100 μM in H2O2 or formaldehyde detection 28 –30 ) to generate a measurable signal in a linear range by using a similar reaction time for both assay formats, the sensitivity of detection for the current TR-FRET assay is much higher than the coupled assay: the coupled assay format is at least 10-fold less sensitive than the TR-FRET format for LSD1 enzyme (compare Figs. 3B and 5) and for substrate needed for generating a quality signal.
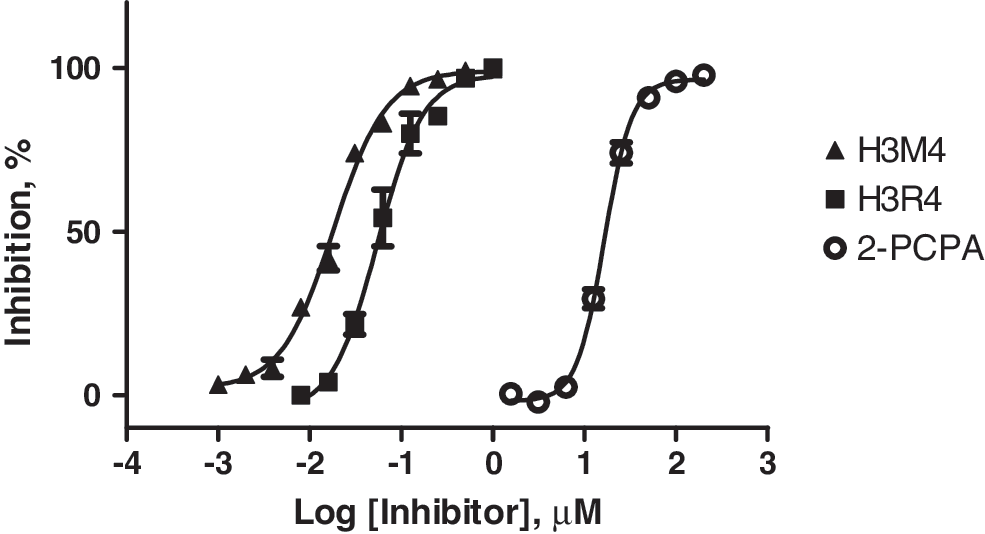
Inhibition profile of known covalent and reversible LSD1 inhibitors as determined in TR-FRET assay (in triplicate, mean±SD; see text for details).
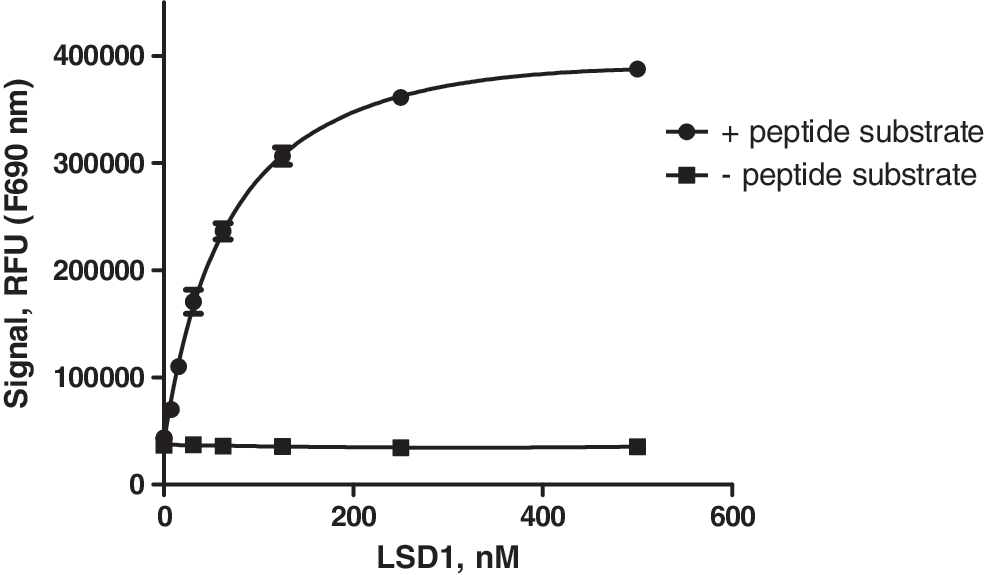
A hydrogen peroxide (H2O2) coupled LSD1 assay measured by the generic Amplex Red detection coupled with horseradish peroxidase (HRP) (in triplicate, mean±SD; see text for details).
The simple and homogeneous TR-FRET assay format is also suitable to find inhibitors using HTS. As a proof of concept, we have tested the TR-FRET assay format in a pilot HTS experiment by using a miniaturized 1,536-well plate format (Table 1). We screened a compound set (approximately 14,000 LMW compounds) against a preparation of LSD1 in duplicate experiments, and the results of this pilot test are shown in Figure 6. The assay showed to be of high quality as judged by a calculated Z′ factor of 0.85 or higher for all plates in the miniaturized 1,536-well format (data not shown) and excellent data repeatability of the assay. Twenty four compounds showed activity of ≥30% inhibition in both the duplicate runs. Two of the 24 hits were also found to be structural analogs of known inhibitors (data not shown). This pilot test demonstrates the usefulness and suitability of the assay in inhibitor screening (Figs. 4 –6).
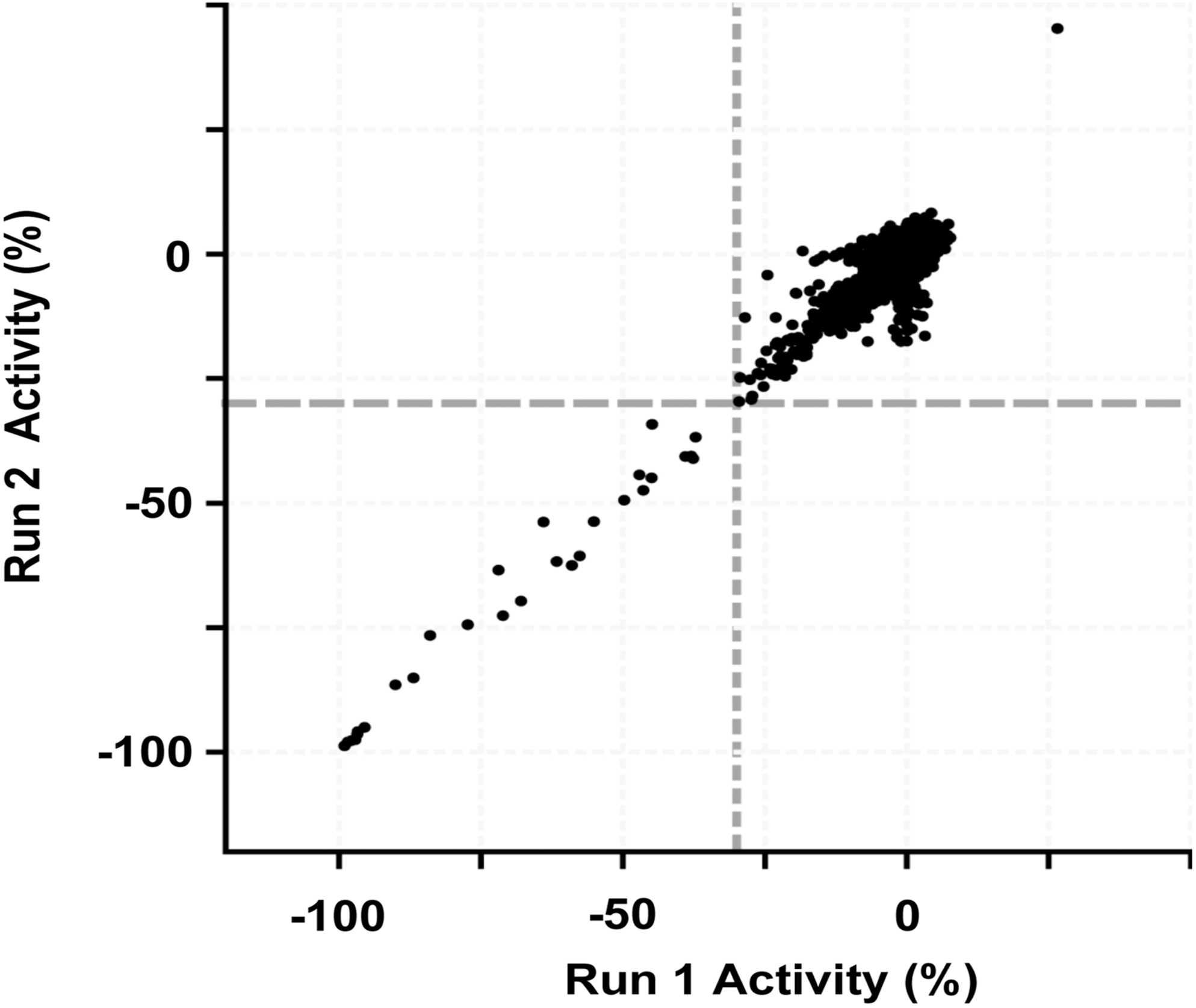
Correlation of screening data from a pilot screen for inhibitors of LSD1 with the TR-FRET assay as described in the text. Approximately 14,000 compounds were independently tested twice (Run 1 and Run 2) at a 10 μM concentration.
Discussion
HMTs and demethylases are two emerging classes of enzymes with important roles in epigenetic changes and are potential biological targets for drug discovery. Most of the currently available assays for histone demethylases suffer from either low throughput (e.g., western blot, ELISA) or sensitivity (e.g., coupled enzyme assays and LC-MS assays). Microfluidic (e.g., LabChip™) and LC-MS detection with improved throughput have also proved to be very useful detection methods for demethylases. However, such throughput or sensitivity limitations not only require more reagent consumption, but also more importantly hinder the use of these methods by raising the lower limit of detection, thus causing unrealistic IC50 shifts of potent inhibitors to higher values, which could mislead SAR studies. Truly high-throughput and sensitive assays for KDMs are needed, and to date, there have been no reports on use of such assays for detection of demethylated peptide products in HTS, albeit assays employing coupling enzymes have been used in HTS against Jumonji demethylase. 31 We collaboratively conceptualized and developed between our labs a TR-FRET assay for H3K4 demethylases where substrate demethylation yields a signal increase, based on a novel and specific anti-H3K4Me0 (unmethylated) antibody labeled with the TR-FRET donor fluorophore—Eu-chelate. The assay is based on the concept of “epitope-unmasking,” wherein an enzymatic reaction leads to the production of an epitope. 32 To our knowledge, the LSD1 assay reported here is the first high-throughput TR-FRET assay using a signal increase mode of detection for histone demethylases.
The TR-FRET signal-assay format for H3K4 site demethylation described here has several advantages. The specificity and selectivity of the employed antibody to the H3K4 substrate is high, as methylation on K4 site shows complete blocking of the TR-FRET signal generation at a broad peptide concentration range, whereas modifications on other methylation sites or unmethylated sites as tested in this study show no appreciable signal generation under the testing condition. The inhibitor peptides (H3M4 and H3R4) do not generate any signal. The sensitivity of the assay is in the low nanomolar range for demethylated peptide substrate and in subnanomolar concentration for LSD1 with the enzyme lot used in our testing conditions. When we compared the current LSD1 TR-FRET assay with a coupled LSD1 assay based on coupled H2O2 detection, we found that the sensitivity of the TR-FRET assay is much higher than the coupled assay. These results demonstrate that the TR-FRET assay format is a sensitive, simple homogeneous assay to enable for inhibitor potency profile and HTS.
It is noteworthy that the current TR-FRET assay configuration is not only suitable for H3K4 site demethylation (such as LSD1/2), but should also be easily adapted to detect other modifications at the H3K4 site. For example, deacetylation reactions of H3K4 can also be detected with a signal increase assay configuration, as recently demonstrated with SIRT1. 33 One limitation of the current assay is that this assay cannot be used to measure higher lysine methylation-state changes, which would be of great interest to the field in understanding the functional consequences of lysine methylation states. Another limitation is due to the detection being site specific; the assay system cannot transfer to measure other lysine methylation changes. However, the anti-H3K4Me0 antibody could be sensitive to phosphorylation at the adjacent threonine site, that is, H3T3 site. 20 Therefore, it is likely that the current assay configuration could be also used for detection of dephosphorylation reaction (via phosphatases) on the H3T3 site. Further, the current TR-FRET assay format can also be adapted into AlphaScreen® format or other similar assay formats. Future investigation along these lines should provide broader applications of this sensitive assay method.
Footnotes
Acknowledgments
The authors thank Ophelia Ardayfio for her assistance in acquiring some of the biotinylated peptides and reagents used in this study, and Sandra Cerruti for her help with the DOE experiments. They also thank Dr. Simon Ng, Janice Giudici-Bouchard, and Dr. Lucille Beaudet for their help in setting up the collaboration and reagent supply, as well as Dr. Dejan Bojanic for his support rendered toward the study.
Disclosure Statement
No competing financial interests exist.