
Abstract
FK506 binding protein12.6 (FKBP12.6) binds to the Ca2+ release channel ryanodine receptor (RyR2) in cardiomyocytes and stabilizes RyR2 to prevent premature sarcoplasmic reticulum Ca2+ release. Previously, two different mouse strains deficient in FKBP12.6 were reported to have different abnormal cardiac phenotypes. The first mutant strain displayed sex-dependent cardiac hypertrophy, while the second displayed exercise-induced cardiac arrhythmia and sudden death. In this study, we tested whether FKBP12.6-deficient mice that display hypertrophic hearts can develop exercise-induced cardiac sudden death and whether the hypertrophic heart is a direct consequence of abnormal calcium handling in mutant cardiomyocytes. Our data show that FKBP12.6-deficient mice with cardiac hypertrophy do not display exercise-induced arrhythmia and/or sudden cardiac death. To investigate the role of FKBP12.6 overexpression for cardiac function and cardiomyocyte calcium release, we generated a transgenic mouse line with cardiac specific overexpression of FKBP12.6 using α-myosin heavy chain (αMHC) promoter. MHC-FKBP12.6 mice displayed normal cardiac development and function. We demonstrated that MHC-FKBP12.6 mice are able to rescue abnormal cardiac hypertrophy and abnormal calcium release in FKBP12.6-deficient mice.
Introduction
FK506 binding proteins (FKBPs) are immunophilins that bind to the immunosuppressive drugs FK506 and rapamycin. 2 FKBP12 (also known as fkbp1a) and FKBP12.6 (also known as fkbp1b) are cytoplasmic proteins that share 85% amino acid identity and a similar tissue distribution. Although FKBP12 is highly expressed in cardiomyocytes, FKBP12.6 is the predominant isoform associated with RyR2. 1,3 –5 Despite their similarities in protein structure, FKBP12 and FKBP12.6 have distinct roles for normal cardiac function. These differences are clearly illustrated in mice with targeted deletion of FKBP12 or FKBP12.6. Mice homozygous for a null mutation in FKBP12 are embryonic lethal due to aberrant cardiac development. 6 In contrast, FKBP12.6-deficient mice display normal cardiac development and are viable. However, once mature, adult FKBP12.6-deficient mice display abnormal cardiac physiology including either enlarged hearts 7 or exercise-induced cardiac arrhythmias and sudden death. 8 Furthermore, our recent study demonstrated that FKBP12 is a key regulator for cardiac voltage-gated sodium channel. 9
RyR is a large tetrameric calcium release channel and is composed of four identical subunits (560 kDa). The majority of channel modulators interact with N-terminal regulatory “foot” region. 1 FKBP12.6 has been shown to regulate RyR2 closure via its interaction with RyR2. 10 –14 Two independent mouse strains deficient in FKBP12.6 had distinctively different abnormal cardiac phenotypes. The first strain (129SvEv/C57 hybrid or 129SvEv inbred background) displayed sex-specific adult cardiac hypertrophy. 7 The second strain (DBA/lacJ inbred background) displayed stress/exercise-induced cardiac sudden death. 8 Interestingly, intracellular calcium release appeared to be altered in both FKBP12.6-deficient mouse models, suggesting that secondary genetic factors may contribute to the final pathogenic outcome. However, there are still many unanswered questions regarding the abnormal cardiac phenotypes in FKBP12.6-deficient mice. It has not been tested whether FKBP12.6-deficient mice in 129SvEv/C57 background can develop exercise-induced cardiac sudden death or whether the cardiac functional defects seen in FKBP12.6-deficient mice (in both strains) are a direct consequence of abnormal calcium handling or an indirect consequence of other altered physiological function(s), such as hypertension 7 or an altered immune system. 15 Resolving these questions will help us to further understand the physiological role of FKBP12.6 in heart function and heart failure. In this report, we determined the physiological impact of cardiomyocyte-restricted overexpression of FKBP12.6 on cardiac function and intracellular Ca2+ release in cardiomyocytes. We demonstrated that αMHC-FKBP12.6 mice were able to rescue abnormal cardiac hypertrophy and Ca2+ release phenotype in FKBP12.6-deficient mice. These data provide further insight into the role of FKBP12.6 in Ca2+ release and cardiac hypertrophy.
Materials and Methods
Generation of αMHC-FKBP12.6 Transgenic and αMHC-FKBP12.6/FKBP12.6–Deficient Mice
FKBP12.6-deficient mice were generated as described previously 7 and were maintained in 129SvEv/C57BL backgrounds. To generate αMHC-FKBP12.6 transgenic mice, an α-myosin heavy chain (αMHC) promoter was used to drive a human FKBP12.6 cDNA (coding region), followed by an SV40 early region transcription terminator/polyadenylation site. A myc epitope tag was added in frame to the 5′ end of FKBP12.6 cDNA. The myc-epitope tag allowed us to distinguish transgenic FKBP12.6 from endogenous mouse FKBP12.6 and FKBP12. αMHC-FKBP12.6 mice were generated as previously described. 10 To generate mice in which FKBP12.6 was only expressed in the myocardium (i.e., FKBP12.6 was not present in any other tissue), we first cross-bred αMHC-FKBP12.6+ mice to FKBP12.6 homozygous mutants (FKBP12.6−/−) to generate mice positive for the transgene (i.e., αMHC-FKBP12.6) and heterozygous for the FKBP12.6 knock-out allele (FKBP12.6+/−). αMHC-FKBP12.6+: FKBP12.6+/− were further cross-bred to FKBP12.6−/− mice to generate αMHC-FKBP12.6+: FKBP12.6−/− mice.
Morphological, Histological, and Echocardiogram Analyses
Cardiac structure and function of mice were also assessed by histological analysis and echocardiograph (ECG) as previously described. 16 In brief, the heart weight to body weight ratio (mg/g) was used as a measure of cardiac hypertrophy. Cardiac tissue samples were fixed in 10% formalin in phosphate-buffered saline (pH 7.4), paraffin embedded, and sectioned (8-μm thickness). Sections were further processed and stained using hematoxylin and eosin for regular cardiac histology. ECG analysis was performed as previously described. 17 Two-dimensional guided M-mode ECG was used to measure left ventricular (LV) diastolic and systolic dimensions (LVEDD and LVESD) and septal and posterior wall thickness. These measures were used to derive fractional shortening and estimated LV mass. Beyond standard measures of cardiac chamber size we calculated h/r, which is an eccentricity index that is derived from the mean wall thickness/radius of ventricular end diastole chamber dimension. LV chamber size, h/r ratio, and LV mass were used as markers for ventricular remodeling. Using pulse wave Doppler, aortic ejection times (ET) were measured to determine velocity of circumferential fiber shortening (Vcf) as follows: Vcf=fraction shortening (FS)/ET.
Real Time Quantitative RT-PCR and Western Blot Analyses
Real-time quantitative PCR and Western blot analyses were performed as previously described. 16,18 The relative expression level was normalized to the reference gene ribosomal protein L7. 16,18 Antibodies against myc-epitope tag and FKBP12 were from Sigma and Affinity BioReagents, respectively.
Electrocardiogram and Exercise Testing in Mice
We studied the possibility of exercise-induced cardiac arrhythmia and sudden death in FKBP12.6-deficient mice (129SvEv/C57B6) as previously described in the assessment of FKBP12.6-deficient mice (DBA/LacJ). 8 In brief, five FKBP12.6-deficient and five age-matched littermate wild-type mice (males at 5 months of age) were anesthetized using intraperitoneal injection of ketamine (50 μg/kg) and xylazine (10 μg/kg), and electrocardiogram (ECG) transmitters were implanted in the peritoneum. ECG recordings of ambulatory animals were obtained using radiotelemetry (Data Sciences International) with transmitters implanted in the peritoneum over 72 h before recordings. Continuous recordings were collected for each mouse, but only ECG complexes with clearly defined onset and termination signals were sampled. Standard criteria were used to measure ECG parameters. 19 QT intervals were measured to the end of the biphasic T wave [Tr+Ts; QTc=QTc=QT/(RR/100)]. 20 For stress tests, mice were forced to run on an inclined treadmill until exhaustion and were then intraperitoneally injected with epinephrine (0.1 or 0.5 mg/kg). Heart rates of ambulatory animals were determined by averaging heart rates over 4 h. The genotypes of the mice were not revealed to the investigator until after all measurements were performed and data were analyzed.
Calcium Spark and Calcium Transient Measurement
Single ventricular myocytes from adult mice (4–6 months old) were prepared as previously described 7 and were incubated with 10 μM Fluo-4 AM (Molecular Probes) for 10 min at room temperature, placed in a recording chamber mounted on an inverted microscope (TE300; Nikon), and perfused for 40 min at room temperature with physiological salt extracellular solution (140 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1.2 mM MgCl2, 10 mM HEPES, pH 7.4, and 10 mM glucose). Fluo-4 fluorescence was recorded using a laser scanning confocal head (Bio-Rad Laboratories) attached to an inverted microscope (TE-300; Nikon) with a plan-apo×60 water immersion objective (1.2 NA.; Nikon). Cells were excited with 488-nm light from a krypton–argon laser and depolarized from −70 mV to −42 mV at the holding potential of −40 mV. Linescan images were recorded using Lasersharp software (Bio-Rad Laboratories) at an interval of 0.833 ms per line. The images were analyzed using LaserPix version 4.2 software (Bio-Rad Laboratories). Fluorescence profiles were constructed by averaging three pixels bisecting a Ca2+ spark for each time point in the scan from the linescan images using LaserPix or a custom developed software.
Results
Assessment of Cardiac Hypertrophy and Exercise-Induced Cardiac Arrhythmia and Sudden Death in FKBP12.6-Deficient Mice
According to previous reports, 7,8 FKBP12.6-deficient mice develop either enlarged hearts after 4 months of age in males or exercise-induced cardiac arrhythmia and sudden death in both sexes depending on the mouse strain backgrounds. To confirm the cardiac hypertrophy phenotype, we systematically compared the size of the hearts in FKBP12.6-deficient males and females (129SvEv/C57B6) at 2 and 5 months of age (Fig. 1A). Consistent with previous findings, 7 hypertrophic hearts were seen in 5-month-old FKBP12.6-deficient males, while in both age groups FKBP12.6-deficient females had normal heart size (Fig. 1A). Despite this apparent cardiac hypertrophic phenotype in male adult mutants, as shown previously, 7 ECG analysis demonstrated normal cardiac function in these mutant males when compared to controls.

FKBP12.6-deficient male mice develop cardiac hypertrophy.
To determine whether this cardiac enlargement in FKBP12.6-deficient males was associated with abnormal cardiac gene expression, we analyzed the expression level of several cardiac markers such as atrial natriuretic factor (ANF), αMHC, βMHC, skeletal α-actin (acta1), sarcoplasmic reticulum Ca2+ ATPase 2a (SERCA2a), and phospholamban (pln). 21 –23 ANF, βMHC, and acta1 are only present in embryonic hearts and are reactivated to persistently higher expression levels in hypertropic hearts. 22 SERCA2a and pln are involved in regulating intracellular Ca2+ homeostasis and contractile function of cardiomyocytes and are normally found to be down-regulated in hypertrophic and failing hearts. 21,23 Intriguingly, although cardiac hypertrophy was seen in FKBP12.6-deficient adult male, mRNA levels of these cardiac hypertrophy markers were not elevated (Fig. 1B). This observation suggests that FKBP12.6-mediated signaling is linked to physiological, rather than pathological hypertrophy, 24,25 consistent with FKBP12.6-deficient mice never displaying heart failure, a common end stage for the pathological hypertrophic heart.
To determine whether FKBP12.6-deficient males also develop exercise-induced cardiac arrhythmia and sudden death in addition to hypertrophy, we compared conscious ECG parameters in these mutant mice and male littermates (5–6 month old) (Fig. 2A and 2B). Continuous ECG recordings were collected from each mouse. No significant difference was observed in RR intervals (wild type: 94.0±7 ms; FKBP12.6−/−: 89.1±7 ms), PR intervals (wild type: 34.1±1.9 ms, n=5; FKBP12.6−/− : 33.1±2.4 ms, n=5, p>0.05), QRS duration (wild type: 16.7±1.7 ms, n=5; FKBP12.6−/−: 16.0±1.5 ms, n=5, p>0.05), rate corrected QT intervals (QTc; wild type: 59.2±4.2 ms, n=5; FKBP12.6−/− : 57.8±6.0 ms, n=5, p>0.05), or resting heart rate (wild type: 615±24 bpm; FKBP12.6−/− : 642±29 bpm, n=5, p>0.05). Neither FKBP12.6-deficient nor wild-type mice displayed arrhythmia or syncope during ECG probe implantation or under sedentary conscious conditions.
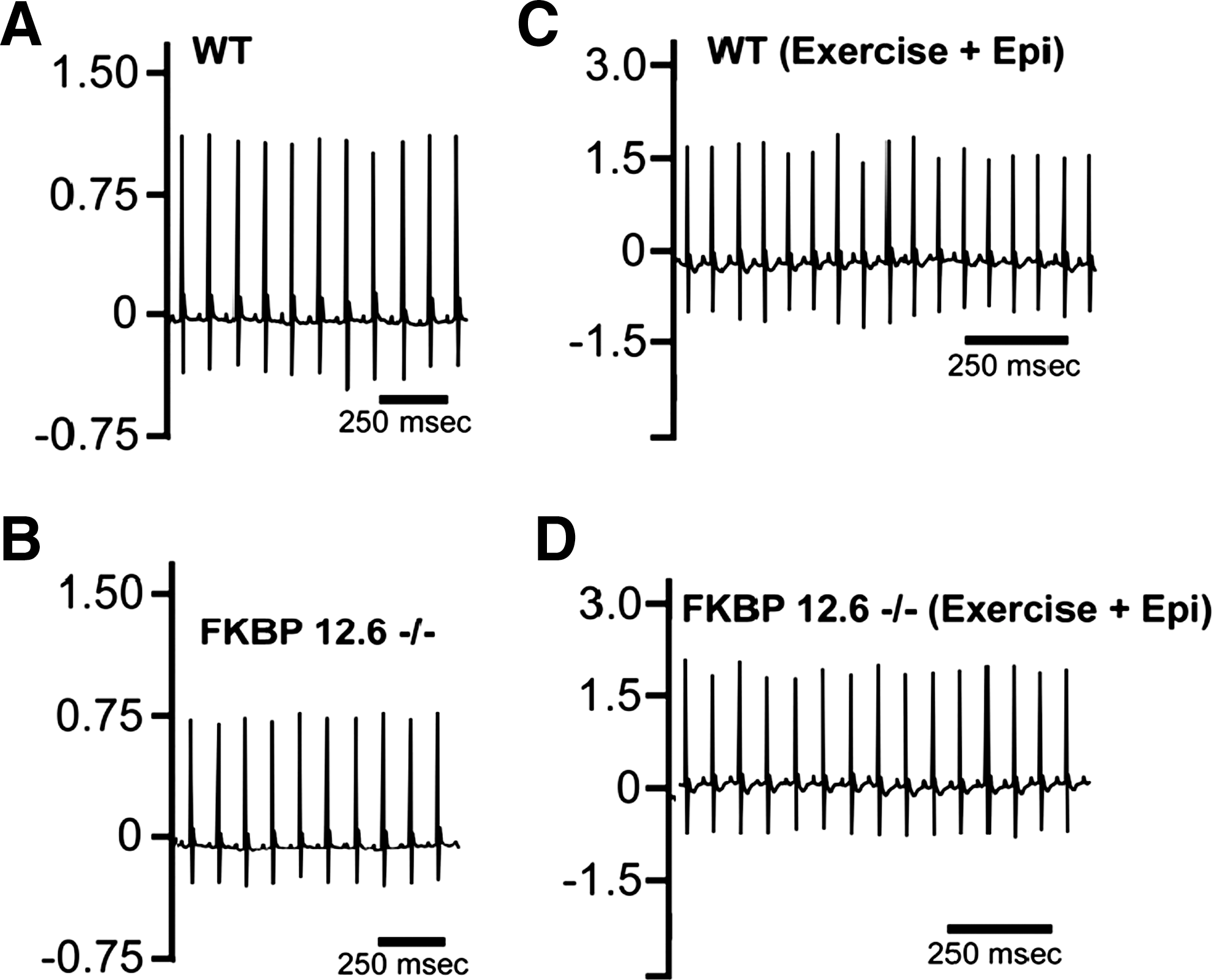
Assessment of exercise induced arrhythmia and sudden death in FKBP12.6-deficient males. ECG traces of littermate control wild-type
To test the FKBP12.6-deficient male for exercise and stress-induced arrhythmia (Fig. 2C and 2D), we subjected the mutant mice and wild-type controls to a strenuous exercise protocol followed by intraperitoneal injection of epinephrine as previously described. 8,20 While both groups of mice had elevated heart rates following exercise, no syncope or polymorphic ventricular arrhythmia were observed in either group (n=5 mice/each genotype). Following intraperitoneal injection of epinephrine, we did not observe differences between the two groups of mice. Again, both groups had elevations in heart rate, but neither FKBP12.6-deficient mice nor wild-type littermates displayed syncope or polymorphic ventricular arrhythmia (n=5 mice/genotype). These observations indicate that the 129SvEv/C57B6 strain of FKBP12.6-deficient mouse does not develop exercise-induced arrhythmia and sudden death.
Generation and Analysis of MHC-FKBP12.6 Transgenic Mice
We generated transgenic mice in which FKBP12.6 expression was driven by a cardiomyocyte-specific alpha myosin heavy chain (αMHC) promoter (Fig. 3A). The αMHC promoter has a transient burst of activity in embryonic heart around E9.5–10.5. This cardiac specific promoter is reactivated during early postnatal life and remains persistently high into adulthood. 26 Five transgenic lines carrying the MHC-FKBP12.6 transgene were generated with similar FKBP12.6 expression levels and with normal cardiac development and function.
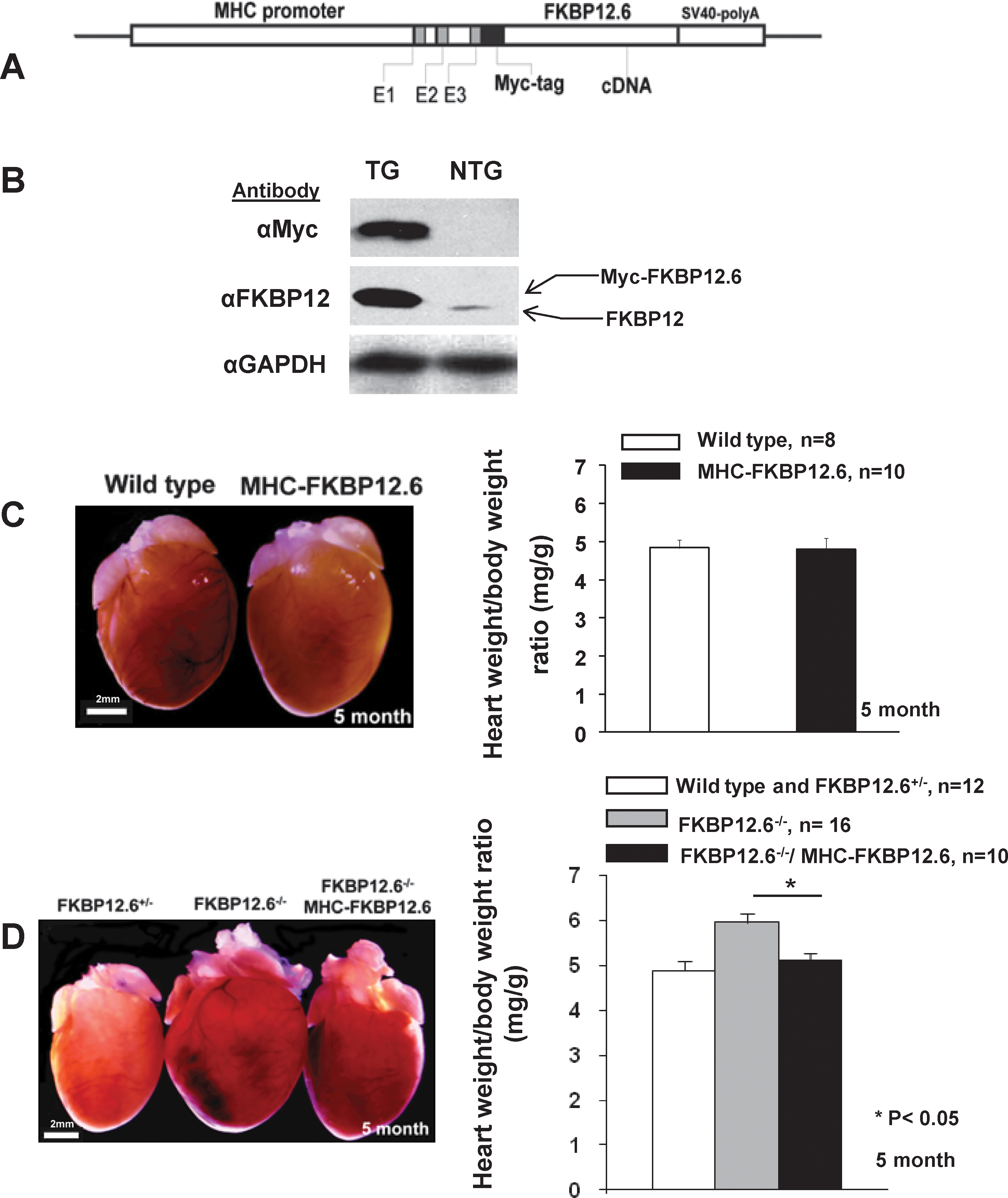
Generation of MHC-FKBP12.6 transgenic mice and αMHC-FKBP12.6/FKBP12.6−/− mice.
Western blot analysis demonstrated that FKBP12.6 expression in MHC-FKBP12.6 hearts was about seven- to ninefold higher than endogenous FKBP12 (Fig. 3B). MHC-FKBP12.6 mice developed normally with no cases of premature death. The heart weight versus body weight ratio in MHC-FKBP12.6 mice was normal in both sexes (Fig. 3). Histological analysis demonstrated that MHC-FKBP12.6 hearts were normal in both ventricles and atria from 1 to 12 months of age. To determine whether MHC-FKBP12.6 mice have altered cardiac function, we performed ECG and Doppler analysis (Table 1). Consistent with our histological observations, MHC-FKBP12.6 (n=6, male, 4 months old) transgenic hearts had normal left ventricular end diastolic and systolic diameter (LVEDD and LVESD), left ventricular mass, percent fraction shortening, and Vcf when compared to littermate controls (n=4). These data suggest that overexpression of FKBP12.6 does not significantly alter cardiac structure and function.
Echocardiograph Analysis
LVEDD, left ventricular diastolic dimension; LVESD, left ventricular systolic dimension; HR, heart rate; h/r, eccentricity index that is derived from taking the mean wall thickness/radius of ventricular end diastole chamber dimension; FS%, percent fraction shortening; Vcf, velocity of circumferential fiber shortening.
Rescuing Cardiac Hypertrophy Phenotype by MHC-FKBP12.6 Transgenic Mice
FKBP12.6 is ubiquitously expressed. One important question was whether the abnormal cardiac hypertrophy seen in FKBP12.6-deficient mice in 129SvEv/C57B6 background was directly caused by the loss of FKBP12.6 expression in cardiomyocytes. In fact, noncardiac defects were also found in FKBP12.6-deficient mice, such as hypertension 7 and renal hypertrophy.* Our next goal was to determine whether this cardiac hypertrophy was secondary to noncardiac or noncardiomyocyte defects in FKBP12.6-deficient mice. To test this, we generated αMHC-FKBP12.6/ FKBP12.6−/− (129SvEv/C57B6) compound mice in which FKBP12.6 transgene was only present in cardiomyocytes. Heart size and cardiac morphology were compared between littermate adult males (5 months old) with different genotypes, αMHC-FKBP12.6/ FKBP12.6−/−, FKBP12.6−/−, and FKBP12.6+/−, and wild type. As shown in Fig. 3D, cardiomyocyte-specific expression of FKBP12.6 could prevent cardiac enlargement in FKBP12.6-deficient adult males. This observation strongly suggested that cardiomyocytes were responsible for the development of cardiac hypertrophy in FKBP12.6-deficient male.
Cardiac Expression of FKBP12.6 Rescues Abnormal Calcium Release
Previously, we had shown that Ca2+ spark and calcium transients were altered in FKBP12.6-deficient cardiomyocytes. 7 These findings were consistent with the probability of RyR2 opening being increased in the absence of FKBP12.6 or pharmacological dissociation of FKBP12.6 from RyR2 channel complex. 8,27 To evaluate Ca2+ release in cardiomyocytes isolated from αMHC-FKBP12.6 transgenic mice, we measured Ca2+ sparks in the cardiomyocytes derived from MHC-FKBP12.6 transgenic hearts. We observed normal Ca2+ release in αMHC-FKBP12.6 cardiomyocytes when compared to wild-type controls (Fig. 4).

Analysis and comparison of Ca2+ sparks in cardiomyocytes.
To determine whether or not the altered Ca2+ release in FKBP12.6-deficient cardiomyocytes was a direct consequence of FKBP12.6 ablation in cardiomyocytes, we measured the characteristics of Ca2+ sparks in cardiomyocytes isolated from αMHC-FKBP12.6/ FKBP12.6−/− (male) and compared them to Ca2+ sparks in cardiomyocytes isolated from sex-matched littermate FKBP12.6−/−, αMHC-FKBP12.6, and wild-type control mice (Fig. 4). These parameters included the frequency and amplitude of the Ca2+ spark, fluorescence magnitude versus average prestimulus fluorescence, full width at half maximum spark size, spark rising time (ms), and half decay time (ms). For all parameters measured, αMHC-FKBP12.6/ FKBP12.6−/− cardiomyocytes had normal Ca2+ sparks. These data indicated that cardiomyocyte-specific expression of FKBP12.6 was able to rescue abnormal calcium release in FKBP12.6-deficient cardiomyocyte (Fig. 4), which further demonstrated a direct association of FKBP12.6 in regulating calcium release channel RyR2 in cardiomyocyte. Taken together, our data provide additional confirmation and insight on the role of FKBP12.6 in regulating cardiomyocyte calcium release and cardiac function.
Discussion
We have carefully analyzed a cardiac hypertrophy phenotype: a total of 173 FKBP12.6 mutants and littermate control mice (88 males and 65 females in two age groups) were analyzed. We also analyzed ECG records of FKBP12.6-deficient mice under resting and extreme-exercise conditions. No exercise-induced cardiac arrhythmia and sudden death were seen in FKBP12.6-deficient mice. We also generated FKBP12.6 transgenic mice and assessed the impact of up-regulated FKBP12.6 expression on cardiac function. For the most part, our data were consistent with a recent finding in an inducible FKBP12.6 transgenic line, 28 except that we did not observe significant alteration in Ca2+ release in the cardiomyocytes isolated from MHC-FKBP12.6 transgenic mice, which could be due to two different transgenic expression systems having been used in the studies. What we observed was the consequence of chronic up-regulation of FKBP12.6, which is different from the temporally controlled up-regulation of FKBP12.6 in the inducible transgenic system. 28 By crossing FKBP12.6-deficient mice to MHC-FKBP12.6 transgenic mice, we demonstrate that cardiomyocyte-restricted overexpression of FKBP12.6 prevents cardiac hypertrophy in FKBP12.6-deficient mice. This observation strongly indicates a direct impact of FKBP12.6 on cardiomyocyte and cardiac function.
The regulatory role of FKBP12.6 in cardiac function and calcium release in cardiomyocytes via RyR2 has been extensively studied in the past decade. It is believed that FKBP12.6 stabilizes the RyR2, and therefore has a direct impact on cardiac excitation–contraction coupling. 1 Depolarization of the sarcolemma and T-tubules in cardiomyocytes triggers contraction by a dynamic process of calcium-induced calcium release. This release is essential to synchronous and coordinated contraction in the heart. Removal of FKBP12.6 from the RyR2 channel complex either genetically (i.e., knock-out) or pharmacologically with a high dose of FK506 produces a “leaky” RyR2 channel, as demonstrated by the increase of channel opening probability in single channel bilayer recording in FKBP12.6-deficient RyR2 8,29,30 and altered calcium release in FKBP12.6-deficient cardiomyocytes (i.e., prolonged Ca2+ spark and increased Ca2+ spark frequency). 7,27,31 Our study using genetically rescued cardiomyocytes confirms these findings and demonstrates the essential role of FKBP12.6 in regulating RyR2 channel function. Given the altered Ca2+ release phenotype in FKBP12.6-deficient mice and the important role of Ca2+-mediated signaling in cardiac hypertrophy, the mouse model may serve as an interesting in vivo model for testing novel compounds that impact on various disease states of the heart.
Footnotes
Acknowledgments
We wish to thank Dr. Shaoliang Jing and Mr. William Carter of Indiana University Mouse Core for their superb assistance in generating MHC-FKBP12.6 transgenic mice, and Drs. Michael Rubart-Von Der Lohe and Pascal Lafontant for comments. This work was supported in part by National Institutes of Health (WS) and Riley Foundation for Children (WS), the NSF of China (81070095 to H.X.) and the NBRP of China (2007CB512100 to H.X.).
Disclosure Statement
No competing financial interests exist.
Abbreviations
*
Zhu Z, Shou W: unpublished observation. Indiana University, Indianapolis, IN, 2004.