
Abstract
Protein–protein interactions are critical molecular determinants of ion channel function and emerging targets for pharmacological interventions. Yet, current methodologies for the rapid detection of ion channel macromolecular complexes are still lacking. In this study we have adapted a split-luciferase complementation assay (LCA) for detecting the assembly of the voltage-gated Na+ (Nav) channel C-tail and the intracellular fibroblast growth factor 14 (FGF14), a functionally relevant component of the Nav channelosome that controls gating and targeting of Nav channels through direct interaction with the channel C-tail. In the LCA, two complementary N-terminus and C-terminus fragments of the firefly luciferase were fused, respectively, to a chimera of the CD4 transmembrane segment and the C-tail of Nav1.6 channel (CD4-Nav1.6-NLuc) or FGF14 (CLuc-FGF14). Co-expression of CLuc-FGF14 and CD4-Nav1.6-NLuc in live cells led to a robust assembly of the FGF14:Nav1.6 C-tail complex, which was attenuated by introducing single-point mutations at the predicted FGF14:Nav channel interface. To evaluate the dynamic regulation of the FGF14:Nav1.6 C-tail complex by signaling pathways, we investigated the effect of kinase inhibitors on the complex formation. Through a platform of counter screenings, we show that the p38/MAPK inhibitor, PD169316, and the IκB kinase inhibitor, BAY 11-7082, reduce the FGF14:Nav1.6 C-tail complementation, highlighting a potential role of the p38MAPK and the IκB/NFκB pathways in controlling neuronal excitability through protein–protein interactions. We envision the methodology presented here as a new valuable tool to allow functional evaluations of protein–channel complexes toward probe development and drug discovery targeting ion channels implicated in human disorders.
Introduction
Voltage-gated Na+ (Nav) channels are heteromeric transmembrane proteins consisting of a pore-forming α-subunit (Nav1.1–Nav1.9) and accessory β-subunits (β1 to β4); these channels are activated by membrane depolarization giving rise to action potentials and providing the basis for excitability in neurons and cardiomyocytes. 6 Recent discoveries indicate that intracellular fibroblast growth factor 14 (FGF14) is a biologically relevant component of the Nav channel macromolecular complex. FGF14 is a member of the intracellular FGFs (iFGFs; FGF11–13), a group of molecules that remain intracellular, are not secreted, and exhibit selective tissue localization. 7 Through a high affinity monomeric interaction with the intracellular C-terminal tail of Nav channel α subunits (Nav1.1–Nav1.9), FGF14 acts as a multivalent molecule that controls neuronal excitability promoting gating, stability, and targeting of native Nav channels to the action potential initiation site. 8 –13 Co-expression of neuronal FGF14 (FGF14-1b isoform) with different Nav channel isoforms results in modulation of Nav current amplitude and of voltage dependence of channel activation and inactivation of a magnitude and direction that depend upon the channel isoform and are distinct compared with any reported effects of other iFGFs on Nav channel function. 8 –19 Furthermore, genetic deletion of fgf14 in rodents impairs neuroplasticity and cognitive function, and single missense mutations of FGF14 in humans results in neurodegeneration, highlighting the functional relevance of FGF14 as a critical component of the Nav channel macromolecular complex. 10,12,20 –23 Although the structure of the FGF14:Nav channel complex, or of any other iFGF:Nav channel complexes, has not been resolved yet, information on critical residues of the iFGFs:Nav channel interface has been inferred from the FGF13 dimer crystal structure. The analysis of crystal packing contacts of the FGF13 dimer combined with mutagenesis experiments has demonstrated the existence of a conserved iFGF monomer interface that is proposed to mediate both homodimerization and Nav channel binding. 9 Point mutations of presumptive hot spots at this interface impair FGF13 regulation of Nav currents and disrupt subcellular targeting and co-localization of FGF14 with native Nav channels at the axonal initial segment (AIS). 9 Overall, the functional significance of FGF14 and the availability of structural information on the iFGFs:Nav channel complex makes the FGF14:Nav channel complex a potential target for proteomics-based discoveries and drug development directed toward regulation of Nav channel function and, ultimately, for treatment of disorders associated with dysregulation and/or mutations of Nav channels (epilepsy, neurodegeneration, pain, or other channelopathies).
As a first step in an FGF14-based medication development, we identified the need for simple and rapid methods for the detection and functional evaluation of protein–channel complexes that could rapidly translate into drug development campaigns targeting ion channels.
Traditional biochemical methods used for protein–protein interaction studies include enzyme-linked immunosorbent assays, surface plasmon resonance, and fluorescence polarization, while the functional effect of protein binding to ion channels has been studied using manual and/or automated patch-clamp electrophysiogy, 24 fluorescence-based methods, 25 or ion flux assays. 26 However, these assays are either relatively low throughput, not optimized for protein–channel complexes (electrophysiology), or costly and time demanding because they require the use of antibodies, high yield of purified proteins, or chemical derivation of the interacting protein pair. Conversely, split-protein reporters have emerged as a powerful methodology for the detection of biomolecular interactions in intact systems. 27 The concept behind this approach relies on the complementation of two separated halves of a monomeric enzyme driven by the assembly of two interacting partners. First utilized with ubiquitin, 28 the approach has been extended to dihydrofolate reductase, 29 β-lactamase, 30 GFP, 31,32 and various luciferase species, such as Renilla luciferase, 33 Gaussia luciferase, 34 and Photinus firefly luciferase. 35 The use of bioluminescence-based assays has become progressively more prominent in recent years. 36 High signal-to-noise ratio, favorable dynamic range, and reversibility of luminescence-based signals have revealed the split-luciferase complementation as a very sensitive assay to detect protein–protein interactions, protein localization, intracellular protein dynamics, and protein activity in real time and in living cells and animals. 35,37
In the present study, we sought to assess the utility of the Photinus firefly split-luciferase complementation assay (LCA) for rapid evaluation of the FGF14:Nav1.6 channel C-tail complex in living cells. Toward this goal, we adapted and optimized the LCA to detect the FGF14:Nav1.6 channel C-tail complex assembly, and further employed the assay to identify critical amino acid residues responsible for the protein–channel interaction and to screen for upstream modulatory elements that alter complex formation. The data presented here support the use of the LCA as an innovative platform for rapid screening of protein–protein interactions within ion channel complexes.
Materials and Methods
Chemicals
D-luciferin was purchased from Gold Biotechnology (St. Louis, MO) and prepared as a 30 mg/mL stock solution in phosphate-buffered saline (PBS); SP600125 (1,9-pyrazoloanthrone) was purchased from EMD Chemicals (San Diego, CA); PD169316 (4-(4-fluorophenyl)-2-(4-nitrophenyl)-5-(4-pyridyl)-1H-imidazole) was purchased from Sigma-Aldrich (St. Louis, MO); and BAY 11-7082 ((E)3-[(4-methylphenyl)sulfonyl]-2-propenenitrile) was purchased from Cayman Chemical (Ann Arbor, MI). The compounds were dissolved in dimethyl sulfoxide (DMSO).
DNA constructs
Mammalian expression vectors coding for N-terminal (pcDNA3.1-V5_HIS TOPO; rapamycin-binding domain [FRB]-N-terminal luciferase fragment [FRB-NLuc]) and C-terminal (pEF6-V5_HIS TOPO; C-terminal luciferase fragment [CLuc-FKBP]) fragments of firefly (Photinus pyralis) luciferase were a gift of Dr. Piwnica-Worms (Washington University, St. Louis, MO). To generate the CLuc-FGF14 construct, FKBP was replaced with neuronal FGF14 (1b isoform) in the CLuc-FKBP fusion vector. CLuc-FGF14 was engineered by polymerase chain reaction (PCR) amplification of the FGF14 open reading frame (nt 1–855) using a 5′ primer containing a BsiWI site up to a linker region and a 3′ primer containing a NotI site and ligated into the CLuc vector. To generate the CD4-Nav1.6-NLuc construct, a chimera carrying the C-terminal fragment of Nav1.6 (amino acids 1763–1976) fused with CD4ΔCtail (amino acids 1–395; gift of Dr. Benedict Dargent, INSERM, France) was similarly replaced with FRB in the FRB-NLuc construct using PCR amplification and ligation into BamHI at the 5′ end and BsiWI at the 3′ end. The choice of using the CD4 chimera fused to Nav1.6 C-tail was based on previous validations of this and other similar constructs in primary hippocampal neurons.
38
–40
Because the N-terminus of the Nav channels is located intracellularly, the fusion of the NLuc fragment to the Nav1.6 C-tail resulted in intracellular reconstitution of the two halves of luciferase. The following primers were used for PCR amplification:
Sense: 5′-CTCGTACGCGTCCCGGGGCGTAAAACCGGTGCCCCTCTTC-3′; Antisense: 5′-GTTTAGCGGCCGCCTATGTTGTCTTACTCTTGTTGACTGG-3′.
Sense: 5′-CGGGGTACCCAAGCCCAGAGCCCTGCCATTTCTGTGGGCTCAGGT3′; Antisense: 5′-CGCGTACGAGATCTGGCACTTGGACTCCCTGACCTCTTTTTGCCT-3′.
The FGF14Y153N/V155N mutant was engineered similarly to CLuc-FGF14 using pQBI-FGF14Y158N/V160N–GFP as a template in the PCR reaction. 9,11 Note that the FGF14Y153N/V155N mutant presented in this study corresponds to the FGF14Y158N/V160N mutant described in previous studies. 9 All constructs were verified by DNA sequencing. The plasmid pGL3 expressing full length firefly luciferase, used for counter screenings of kinase inhibitors, was a gift of Dr. Sarkar (University of Texas Medical Branch [UTMB], Department of Neurology).
Cell culture and transient transfections
HEK293 cells were incubated at 37°C with 5% CO2 in medium composed of equal volumes of Dulbecco modified essential medium (DMEM) and F12 (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. For transfection cells were seeded in 24-well CELLSTAR® tissue culture plates (Greiner Bio-One, Monroe, NC) at 4.5×105 cells per well and incubated overnight to give monolayers at 90%–100% confluency. The cells were then transiently cotransfected with pairs of plasmids or single plasmids using Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. Each plasmid used per transfection per well was 1 μg, unless otherwise indicated. Cells co-transfected with the CLuc-FGF14 and CD4-Nav1.6-C-tail-NLuc constructs (1 μg of each construct per transfection) were used as a positive control; cells co-transfected with CLuc-FGF14 and pcDNA3.1 empty vector (1 μg of each construct per transfection; this pair is referred to in the text as CLuc-FGF14 alone) were used as background luminescence. The same ratio and plasmid DNA amounts were used for the experiments involving the CLuc-FGF14Y153N/V155N and CD4-Nav1.6-C-tail-NLuc complex.
Bioluminescence assays
Forty-eight hours post-transfection, cells were trypsinized (0.25%) for 10 min at 37°C, triturated in a medium, and seeded in white, clear-bottom CELLSTAR μClear® 96-well tissue culture plates (Greiner Bio-One) at ∼105 cells per well in 200 μL of medium. The cells were incubated for 24 h and then the growth medium was replaced with 100 μL of serum-free, phenol red–free DMEM/F12 medium (Invitrogen). In experiments involving protein kinase inhibitors compounds dissolved in DMSO (stock solution=10 mM; intermediate dilutions=0.2 mM, 0.5 mM, 1 mM, 2 mM, 4 mM, 6 mM, 8 mM in DMSO) were added to the final concentration of 1–50 μM in the culture medium; the final concentration of DMSO was maintained at 0.5% and positive control wells were also treated with 0.5% DMSO (treated positive control). Luminescence measurements were performed 1 h after the application of compounds. The reporter reaction was initiated by injection of 100 μL of substrate solution containing 1.5 mg/mL of D-luciferin (final concentration=0.75 mg/mL) dissolved in serum-free, phenol red–free DMEM/F12 medium. Dispensing of the substrate was performed by the Synergy™ H4 Multi-Mode Microplate Reader (BioTek, Winooski, VT). Luminescence readings were initiated after 3 s of mild plate shaking and performed at 2-min intervals for 20 min, integration time 0.5 s. The cells were maintained at 37°C throughout the measurements. Signal intensity for each well was calculated as a mean value of peak luminescence and luminescence measured at two adjacent time points; the calculated values were expressed as percentage of mean signal intensity in the control samples from the same experimental plate.
Molecular modeling
The PDB coordinates for the FGF14 model based on the FGF9 crystal structure were generously provided by Dr. Van Swieten (Department of Neurology, Erasmus Medical Center, Rotterdam). This FGF14 homology model 21 was used as a template. The electrostatic surface representation and FGF14Y153N/V155N mutations were carried out in silico within the University of California San Francisco (USCF) Chimera suite 41 applying the Richardson rotamers algorithm. 42
Measurement of cell viability
The number of viable transfected cells after treatment was estimated by CyQUANT® Cell Proliferation Assay Kit (Invitrogen) according to manufacturer's instructions after incubation with 0.5% DMSO or appropriate kinase inhibitors. Fluorescence was measured using the Synergy™ H4 Multi-Mode Microplate Reader (excitation λ=485 nm, emission λ=528 nm). Cell viability was expressed as percent mean fluorescent signal intensity in the control samples from the same experimental plate.
Data analysis
Statistical values are given as mean and standard error of the mean (mean±SEM), unless otherwise specified. The following control groups were used: (i) CLuc-FGF14+CD4Nav1.6-NLuc, untreated (untreated positive control); (ii) CLuc-FGF14+CD4Nav1.6-NLuc, treated with 0.5% DMSO (treated positive control); (iii) CLuc-FGF14 alone, untreated (reference background); (iv) CLuc-FGF14+CD4Nav1.6-NLuc treated with 50 μM BAY 11-7082 (negative control and background). In the experiments involving the FGF14Y153N/V155N mutant, untreated control was used for comparison. Initial experiments were performed to evaluate the sensitivity of the assay to 0.5% DMSO by comparing untreated positive controls to treated positive controls (n=4, t-test, p=0.262). In the experiments evaluating the effect of kinase inhibitors DMSO treated controls were used for comparison.
Statistical parameters of assay performance were calculated according to the following formulas:
where δp and δn are standard deviation of the positive group p and the negative control group n, and μp and μn are the mathematical means of the two groups, respectively; S:B, signal to background; S:N, signal-to-noise; and SW, signal window. The positive control group for assay performance evaluation was CLuc-FGF14+CD4Nav1.6-NLuc treated with 0.5% DMSO and the negative control was CLuc-FGF14+CD4Nav1.6-NLuc treated with 50 μM BAY 11-7082.
Dose–response curves and IC50 for each compound were obtained by fitting the data with a nonlinear regression:
where x is log10 of the compound concentration in M, x 0 is the inflection point (IC50), A 1 is the initial strength, A 2 is the offset, and p is the Hill slope.
The adjusted coefficient of determination between number of plated cells or amount of transfected cDNA plasmid and luminescence signal (Supplementary Fig. S1; Supplementary Data are available online at
where RSS is residual sum of square, TSS is total sum of square, df total is the degrees of freedom n−1 of the estimate of the population variance of the dependent variable, and dferror is the degrees of freedom n−p−1 of the estimate of the underlying population error variance.
The statistical significance (p<0.05) of observed differences among groups was determined by Student's t-test, one-way ANOVA with post hoc Dunnett's, Kruskal–Wallis one-way ANOVA on ranks with post hoc Dunn's method, or ANOVA using Sigma Stat software (Jandel Inc., San Jose, CA). The ANOVA values were not statistically different from each other and no post hoc analysis was required. The choice of ANOVA (parametric) versus Kruskal–Wallis (nonparametric) tests was based on normality and equal variance analysis performed with Sigma Stat. Fitting and graphs were generated in Origin Software (Origin Lab Corporation, Northampton, MA).
Western blotting
Transfected HEK293 cells treated with compounds were washed with PBS and lysed in buffer containing 20 mM Tris-HCl, 150 mM NaCl, and 1% NP-40. Protease inhibitor cocktail (set 3, Calbiochem, La Jolla, CA) was added immediately before cell lysis. Cell extracts were collected, sonicated for 20 s, and centrifuged at 4°C, 15,000 g for 15 min adding 2 × sample buffer containing 50 mM tris(2-carboxyethyl)phosphine. Mixtures were heated for 10 min at 65°C and resolved on 4%–15% polyacrylamide gels (BioRad, Hercules, CA). Resolved proteins were transferred to PVDF membranes (Millipore, Bedford, MA) for 1.5 h at 4°C and blocked in tris-buffered saline with 3% nonfat dry milk and 0.1% Tween-20. Membranes were then incubated overnight in blocking buffer containing the anti-luciferase goat polyclonal antibody (Promega, Madison, WI) or anti-calnexin rabbit polyclonal antibody (Cell Signaling Technology, Danvers, MA). Washed membranes were incubated with donkey anti-goat or goat anti-rabbit-horseradish peroxidase (1:5000) and visualized with ECL Advance Western Blotting Detection kit (GE Healthcare, Piscataway, NJ); protein bands were visualized using FluorChem® HD2 System and analyzed with AlphaView 3.1 software (ProteinSimple, Santa Clara, CA).
Results
LCA detection of the FGF14:Nav channel C-tail complex
To develop reliable methods to study protein–channel complexes, we initially set out to determine whether the FGF14:Nav channel C-tail complex assembly was detectable in live cells. We adapted the LCA to detect the assembly of FGF14 and the C-terminus tail of the Nav1.6 channel in HEK293 cells. In this assay, two constructs bearing the complementary N-terminus (NLuc) and C-terminus (CLuc) fragments of the firefly luciferase 35 were fused, respectively, to a chimera of the CD4 transmembrane segment and the C-tail of Nav1.6 38 (CD4-Nav1.6-NLuc) or FGF14 (CLuc-FGF14) (Fig. 1A) and transiently expressed in HEK293 cells, following a 3-day experimental scheme (Table 1). Co-transfection of CD4-Nav1.6-NLuc and CLuc-FGF14 constructs in HEK293 cells led to ∼50-fold increase in the luminescence signal generated after addition of the substrate, D-luciferin (Fig. 1B), compared to background (CLuc-FGF14 alone, Fig. 1C). The luminescence signal reached a steady-state maximal value after 12–15 min that persisted to the end of each experiment (20–30 min), indicating a robust assembly of the FGF14:Nav1.6 complex in live cells. Optimal amounts of transfected plasmid DNA and number of transfected cells seeded per well prior to luminescence measurements were determined as part of the assay development effort (Table 1, Supplementary Fig. S1). The level of luminescence correlated with the amount of transfected plasmid DNA (R 2=0.96) and the number of cells plated (R 2=0.99). A total of 2 μg of DNA (1 μg per construct) and a cell seeding density of 1.5×105 cells/well (96-well plate) produced the strongest luminescence signal (Supplementary Fig. S1). Increasing the total amount of transfected DNA above 2 μg resulted in lower luminescence signal (data not shown), presumably due to cell toxicity. Based on these results, we concluded that LCA is a useful tool for rapid evaluation of ion channel macromolecular complexes in live cells.
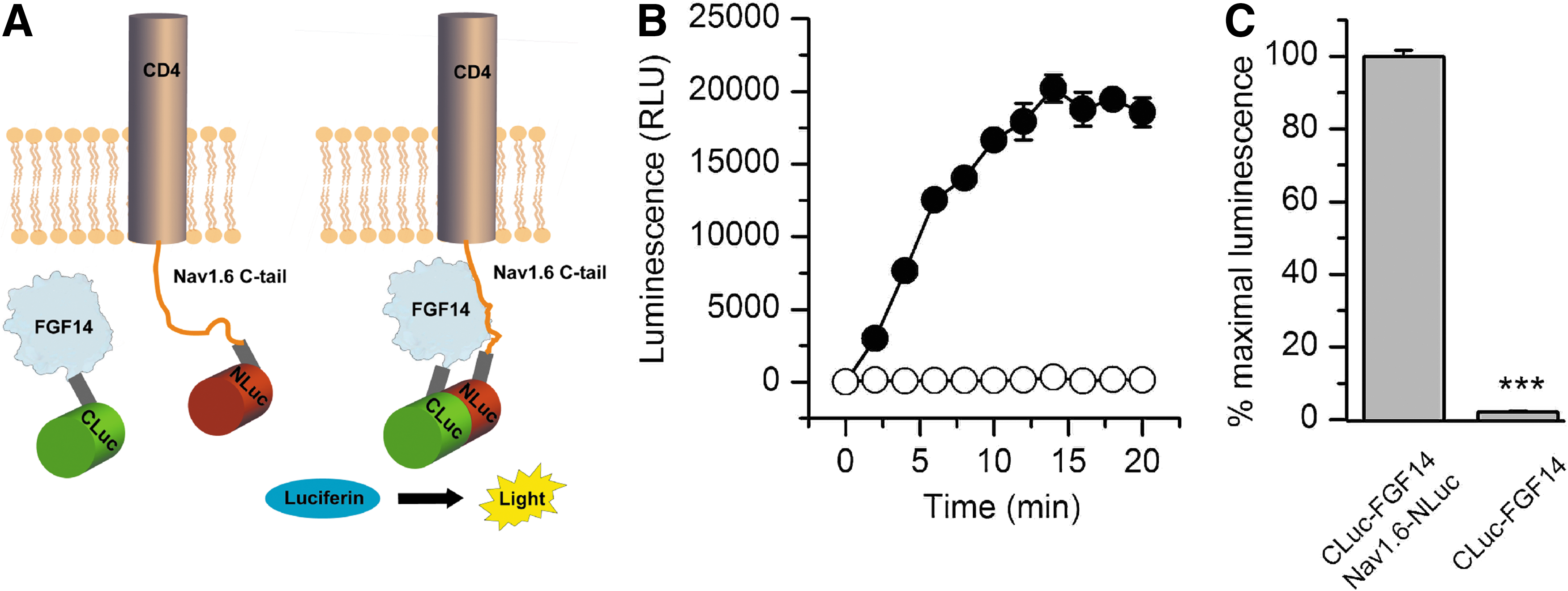
Bioluminescence detection of the FGF14:Nav1.6 C-tail complex in live cells using the split-luciferase complementation assay.
Bioluminescence Assay Protocol
Step Notes
1. Greiner white-walled 24-well plate.
2. 37°C and 5% CO2.
3. Greiner white-walled 96-well plate.
4. Stock concentration of compounds is 0.2–10 mM; 0.5% DMSO control.
5. 37°C and 5% CO2.
6. Detection of complementation of the luminescent protein pairs.
7. Synergy H4 Multi-Mode Microplate Reader; integration time 0.5 s.
PMT, photomultiplier tube.
LCA validation of protein–channel interface hot spots
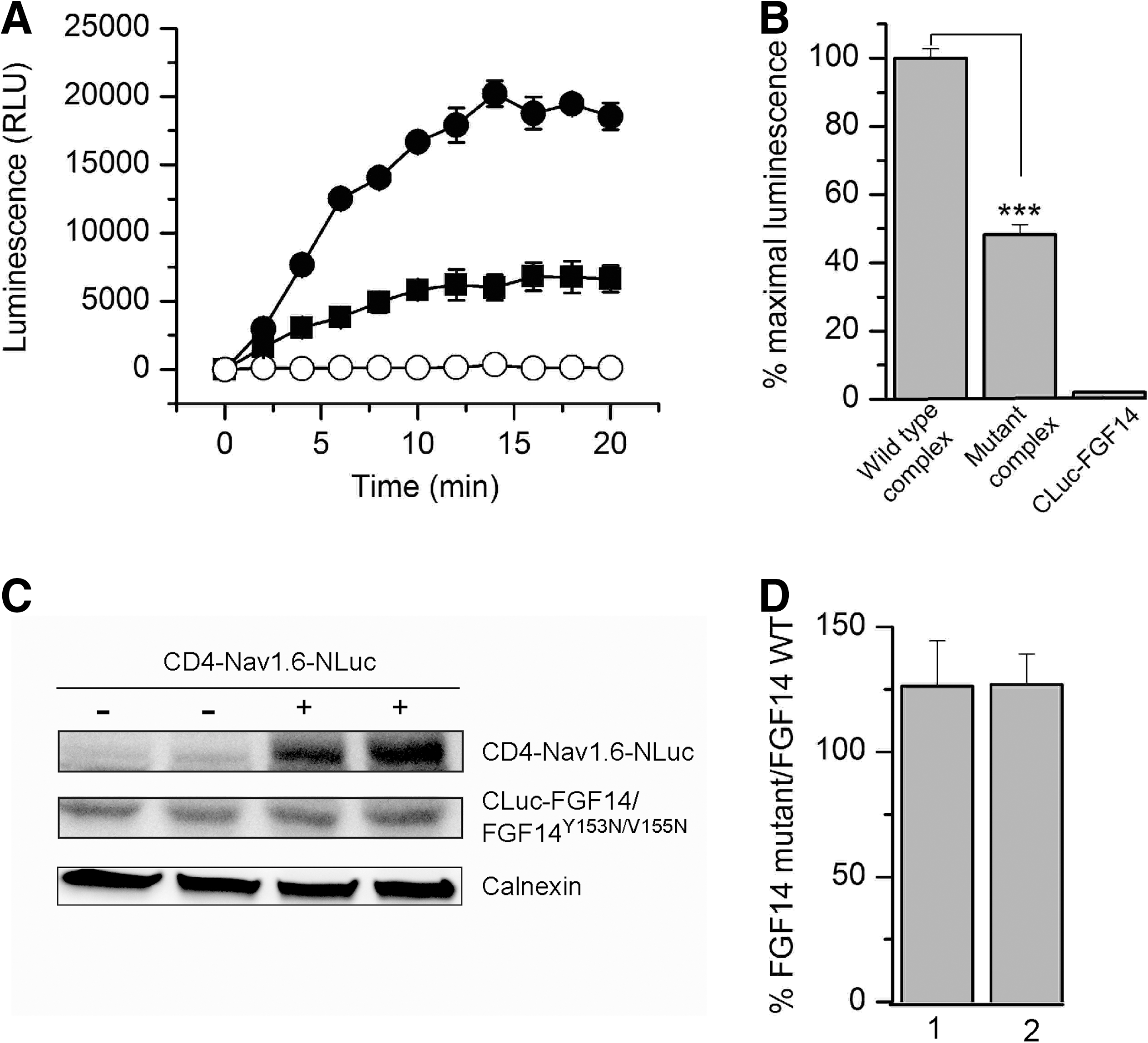
To evaluate whether LCA would be suitable for in-cell validation of hot spots at protein–channel interfaces, we showed in silico mutations of critical residues at the predicted FGF14:Nav channel interface. FGF14 and other iFGFs bind to the Nav channel C-tails as monomers and this homodimerization interface is also responsible for Nav channel binding. 9 Residues Y153 and V155 in FGF14 and the corresponding residues in all iFGFs have been shown to be critical for Nav channel binding. 9 Thus, we posited that that these residues could be hot spots at the FGF14:Nav channel interface. To visualize the effect of the previously studied FGF14Y153N/V155N double mutation 9 at the potential FGF14:Nav channel interface, we carried out in silico the Y153N and V155N mutations (Fig. 2). Mutating Tyr153 to Asn reduces the net negative charge due to the OH group from the Tyr side chain (Fig. 2A, B), additionally modifying the β8-β9 loop surface due to the replacement of the cyclic bulky Tyr ring with the less bulky Asn side chain (Fig. 2D). Mutating the Val155 to Asn modifies further the cavity adjacent to Tyr153 (Fig. 2D). Simultaneously mutating Tyr153 and Val155 to Asn reduces the overall negative charge and smoothes the predicted FGF14:Nav channel interface at the level of the β8-β9 strands loop, where Tyr153 and Val155 are positioned. We utilized the LCA to determine whether these predicted structural alterations in the channel-binding interface would result in decreased assembly of the FGF14:Nav1.6 complex. The complementation of the FGF14Y153N/V155N:Nav1.6 complex was measured over time (Fig. 3A) and was significantly reduced in comparison to the wild type complex (Fig. 3B, 48.3±3%, n=4, Student's t-test, p<0.001). However, since mutations are known to induce protein misfolding and degradation, 43 it was necessary to rule out changes in the protein expression level induced by the Y153N/V155N mutations as a mechanism for the reduced assembly of the mutated complex. To this end, we performed additional control experiments in which expression of CLuc-FGF14 and CLuc-FGF14Y153N/V155N was confirmed by Western blot analysis (Fig. 3C). The expression level of FGF14Y153N/V155N was comparable to FGF14 wild type, regardless of the presence or absence of CD4-Nav1.6-NLuc (Fig. 3C, D). Furthermore, FGF14Y153N/V155N did not significantly affect the expression level of CD-Nav1.6-NLuc (108±13% compared to CD-Nav1.6-Nluc+FGF14 wild type, n=3, paired t-test, p=0.49). These results confirm that the nonproductive assembly of the FGF14Y153N/V155N:Nav1.6 channel C-tail was not attributable to significant differences in the protein expression levels, but was rather consistent with structural changes at the level of the protein–protein interaction interface.
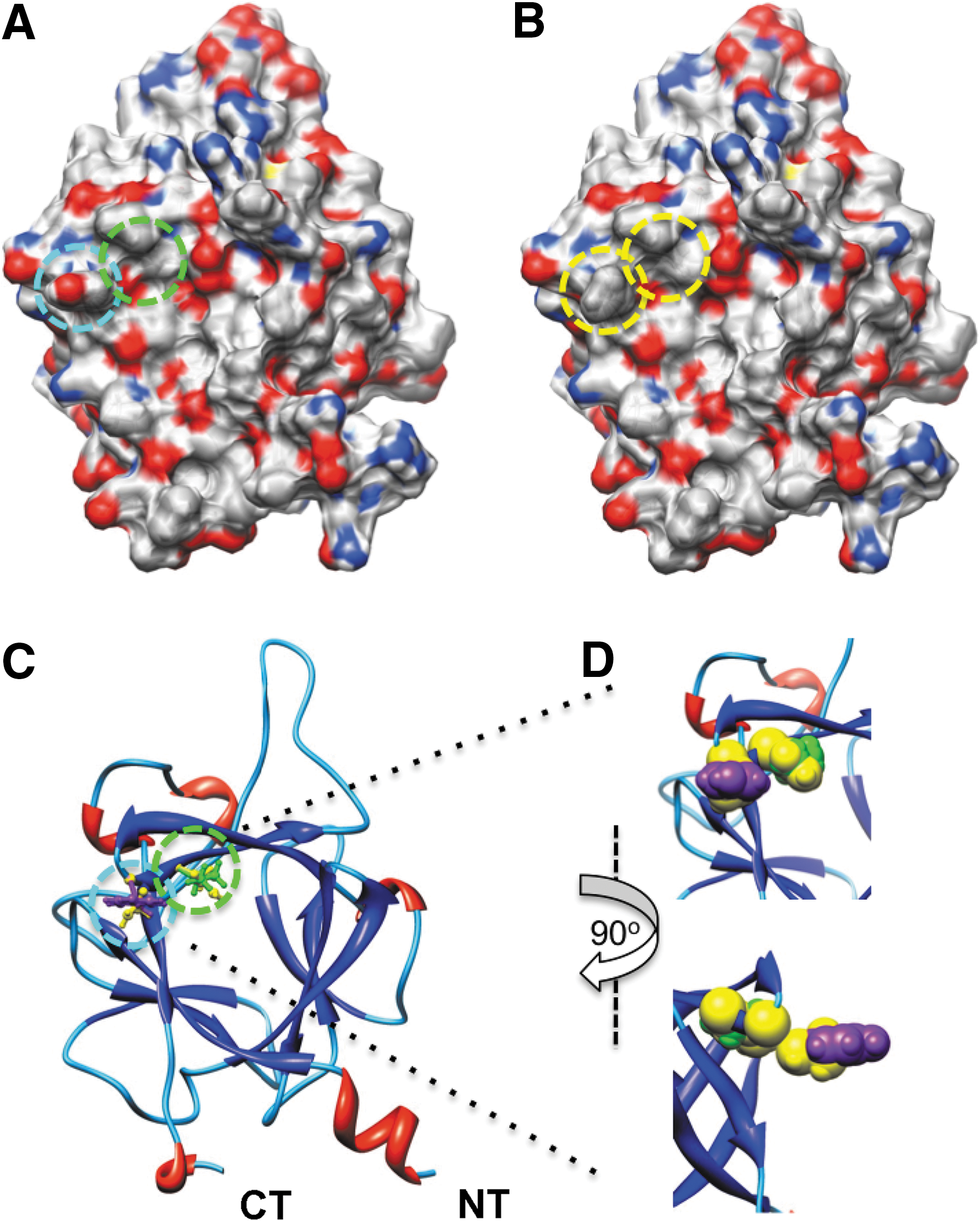
In silico model of the FGF14Y153N/V155N mutations. Electrostatic surface representation of FGF14 wild type
LCA validates in silico predictions of the structure-function studies.
In-cell dynamic regulation of the FGF14:Nav1.6 C-tail complex
Having established that the LCA provides a rapid method for in-cell evaluation of critical residues at protein–protein interfaces, we next explored the dynamic regulation of the FGF14:Nav1.6 C-tail complex by intracellular signaling pathways, focusing on the role of kinases. Kinases are key regulators of neuronal excitability,
44,45
and phosphorylation is a posttranslational modification well known to affect macromolecular complexes. In the brain Nav channels are phosphorylated at multiple intracellular sites,
44,46
and these posttranslational modifications result in modulation of channel gating and trafficking with effects on neuronal excitability.
47
Interestingly, FGF14 sequence motif analysis (
To test this hypothesis, we evaluated the effect of selective kinase inhibitors on the FGF14:Nav1.6 channel C-tail complex assembly. We chose chemical inhibitors of the mitogen-activated protein kinase (MAPK) p38 and the IκB kinase (IKK), a repressor of the nuclear factor κB (NFκB). Both MAPK/p38 and IKK have been directly or indirectly linked to iFGFs. In earlier studies, FGF12 was identified as a binding partner of islet brain 2 (IB2/JIP2), a scaffold protein of MAPKp38δ, 46 and IKK has been recently found at the AIS in a complex with iFGFs. 48 To determine the role of these kinases in modulating the FGF14:Nav1.6 channel C-tail complex assembly, HEK293 cells, co-transfected with CLuc-FGF14 and CD4-Nav1.6C-tail-NLuc, were exposed to the MAPKp38 inhibitor PD169316, the IKK inhibitor BAY 11-7082, the c-JNK (c-JNK terminal kinases) inhibitor SP600125, or vehicle (0.5% DMSO) for 1 h prior to the assay. The c-JNK cascade is another branch of the MAPK pathway, 49 and SP600125 was used as an internal control for specificity. Upon substrate addition, luminescence from various experimental groups was measured up to 20 min (Fig. 4A). Compared to DMSO, treatment with PD169316 or BAY 11-7082 significantly reduced the FGF14:Nav1.6 assembly to 54±7.7% (n=3, p<0.01) and 52±3.5% (n=3, p<0.001), respectively, while no significant changes in luminescence were observed upon SP00125 treatment (104±6.6%, n=3, p=0.53, Fig. 4B). These data suggest that activation of the MAPKp38 and the IκB/NFκB pathways in vivo might promote assembly and stability of the FGF14:Nav channel complex. Whether these effects result from a direct phosphorylation of the complex remains to be determined.
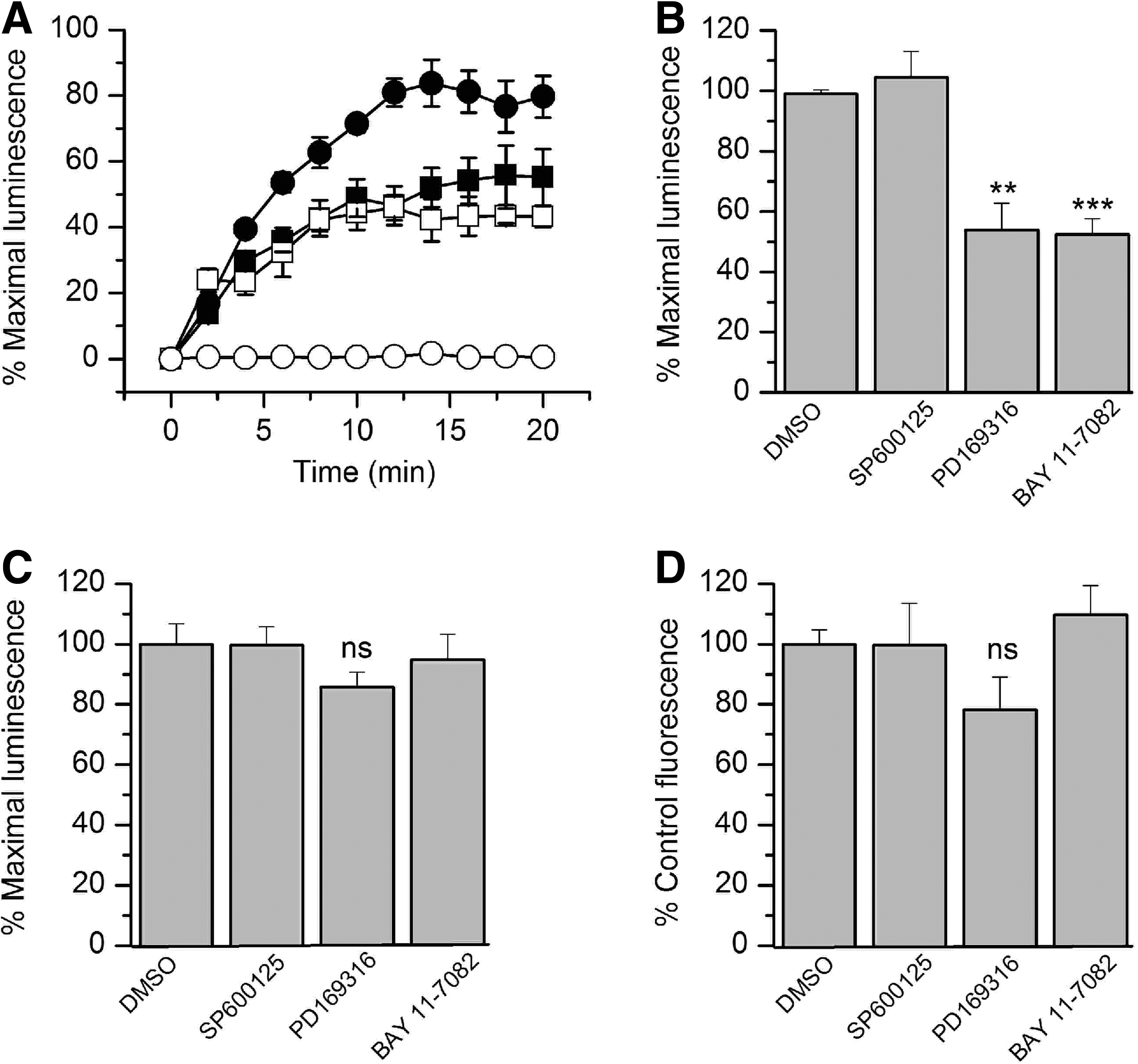
The FGF14:Nav1.6 functional complementation is regulated by specific kinase inhibitors.
Reports indicate chemical interference of compounds with the enzymatic activity of various species of luciferase. 50,51 To rule out that the observed results were induced by the effect of the compounds on the enzymatic activity of luciferase, the kinase inhibitors were screened against HEK293 cells transfected with full length firefly luciferase. Cells were exposed to either PD169316 (10 μM), BAY 11-7082 (10 μM), SP600125 (50 μM), or DMSO for 1 h; none of these compounds had a significant effect on firefly luciferase activity compared to DMSO-treated controls (Fig. 4C; PD169316, 86±5%, p=0.11; BAY 11-7082, 95±9%, p=0.64; SP600125, 112±6.2%, p=0.21; for all groups n=3). Furthermore, we performed additional control experiments to rule out other potential artifacts including reduced cell viability or altered expression levels of the split constructs. Cell viability across experimental conditions was determined by the CyQuant fluorescence-based proliferation assay, and revealed no significant differences across conditions (Fig. 4D; PD169316, 78±11%, p=0.14; BAY 11-7082, 109.7±9.7%, p=0.42; SP600125 99.6±13.9%, p=0.98; for all groups n=3). Western blot analysis confirmed that the CD4-Nav1.6-NLuc and CLuc-FGF14 expression levels were not affected by treatments with PD169316 (10 μM), BAY 11-7082 (10 μM), or SP600125 (50 μM) compared to DMSO-treated control cells (Fig. 5). Overall, these results indicate that the FGF14:Nav1.6 channel C-tail assembly is controlled by the activity of specific kinases. This mechanism might contribute to a complex, yet unknown, regulation of neuronal excitability through protein–protein interaction and provide a platform to develop chemical interventions against Nav channel–related diseases based on phosphoproteomic networks.
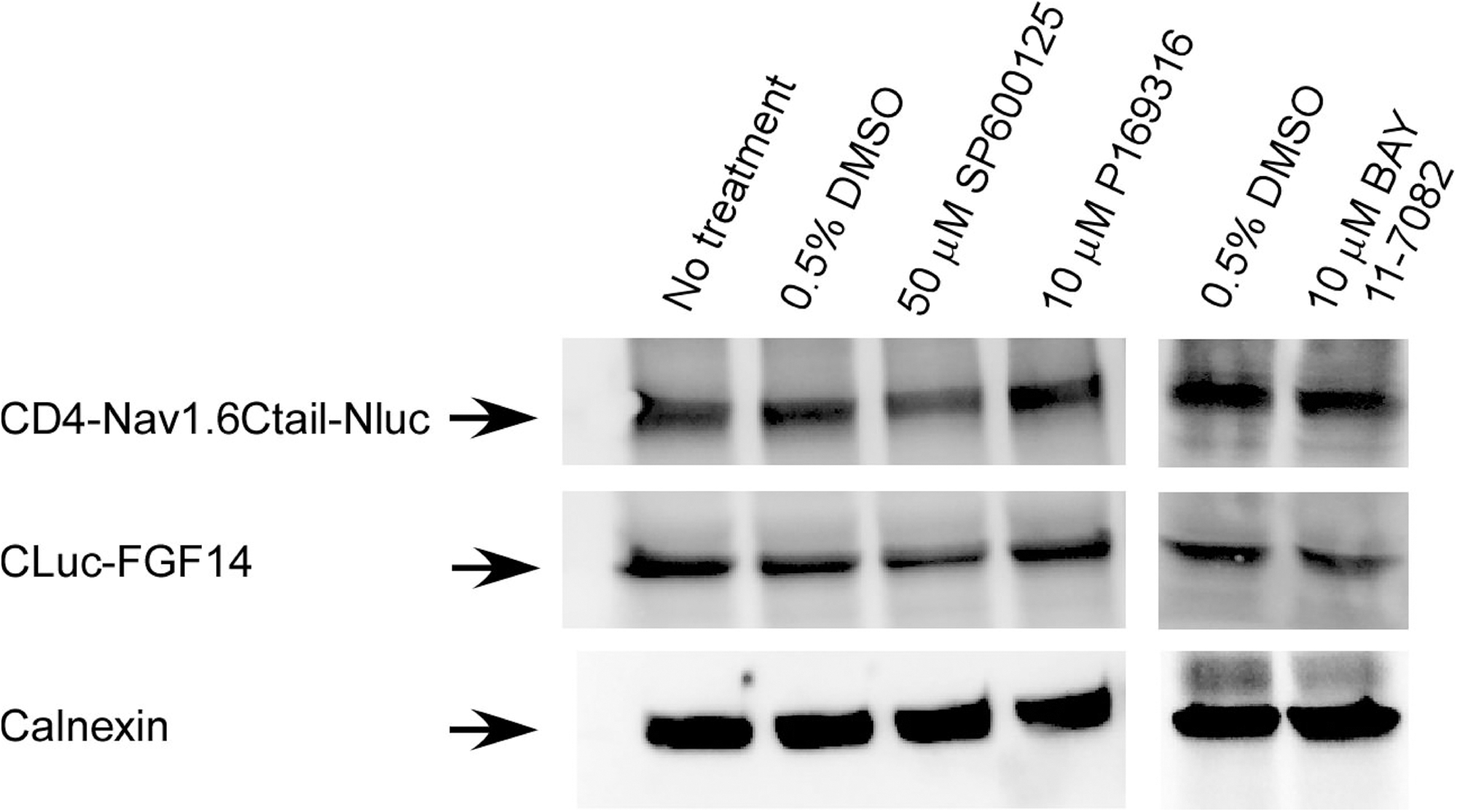
Effect of kinase inhibitors on protein expression levels. Representative example of Western blots of whole-cell extracts (equal amount of protein per lane) from cells transfected with CLuc-FGF14+CD4-Nav1.6-NLuc and treated for 1 h with the indicated compounds (total number of experiments=3). Western blots were probed with a polyclonal anti-luciferase antibody; immunodetection of calnexin is used as loading control.
In-cell dose responses using LCA
Finally, we determined the use of the LCA as a rapid tool for in-cell pharmacology. HEK293 cells transiently transfected with CLuc-FGF14+CD4Nav1.6-NLuc were exposed to a range of concentrations (1–50 μM) of either PD169316 or BAY 11-7082, or to DMSO (0.5%) for 1 h; maximal luminescence for each experimental group was used to construct dose–response curves. Fitting with nonlinear regression yielded comparable IC50 values of 8.9±0.93 μM and 8.8±0.85 μM for PD169316 and BAY 11-7082, respectively (Fig. 6). Notably, the IC50 values of both kinase inhibitors were consistent with those reported in other in-cell studies. 52,53 In an effort to evaluate assay reproducibility, Z′-factor, signal-to-background, signal-to-noise ratio, and signal window 37,54,55 were determined by comparing luminescence values in CLuc-FGF14+CD4Nav1.6-NLuc cells treated with 0.5% DMSO (positive control) versus BAY 11-7082 (50 μM; negative control and/or background), as illustrated in Figure 7. Exposure of cells to 50 μM BAY 11-7082 did not affect cell viability (132.4±18.3% compared to DMSO control, n=3, t-test, p=0.25). Overall, these results indicate that upon optimization to 384- or 1536-well plate format LCA would be valuable for rapid evaluation of compound potency in cells and might be used to expedite hit validation in high-throughput screening campaigns.
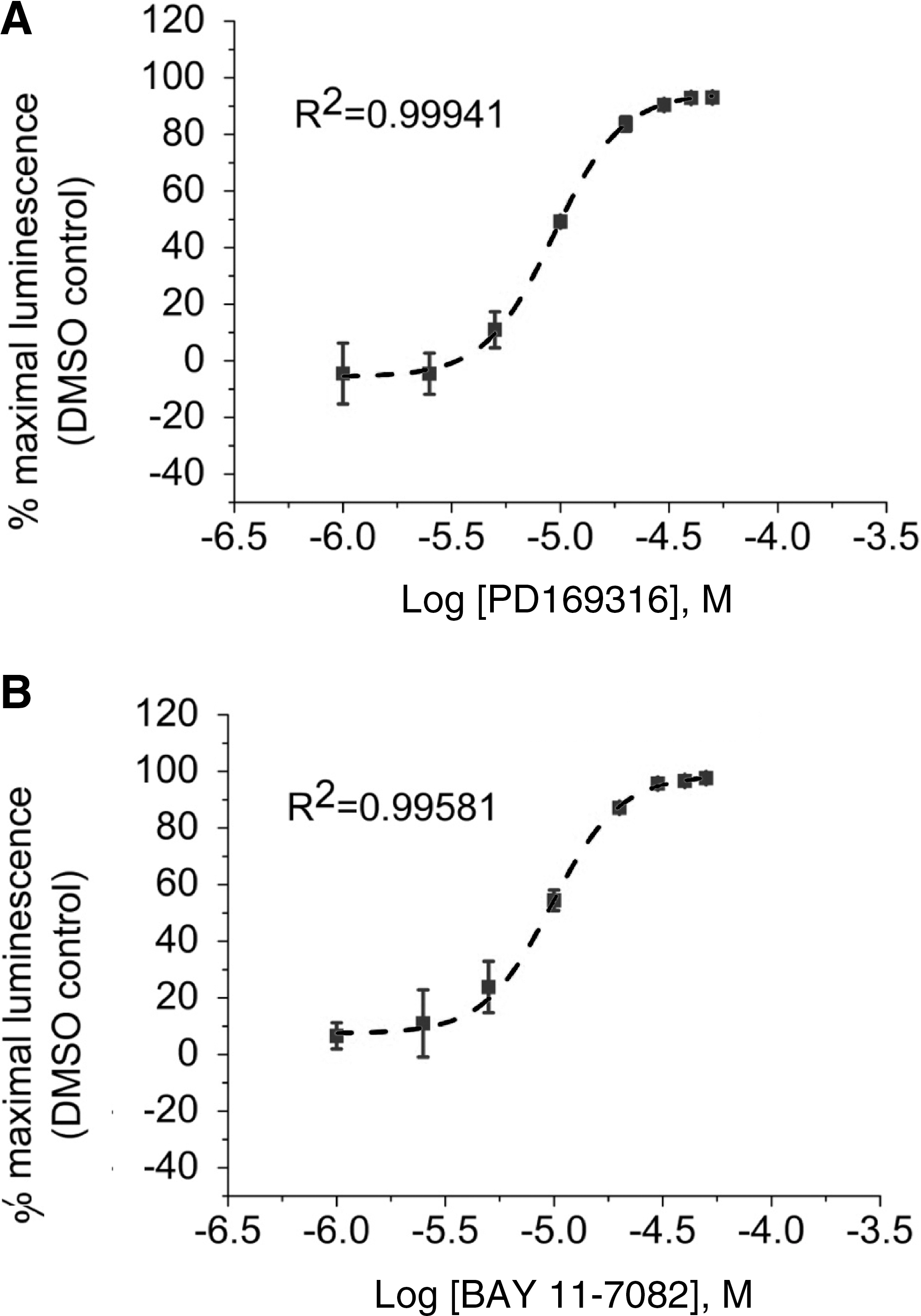
Evaluation of the potency of selective kinase inhibitors. HEK293 cells were transiently transfected with CLuc-FGF14 and CD4-Nav1.6-NLuc and treated with the indicated concentrations of PD169316
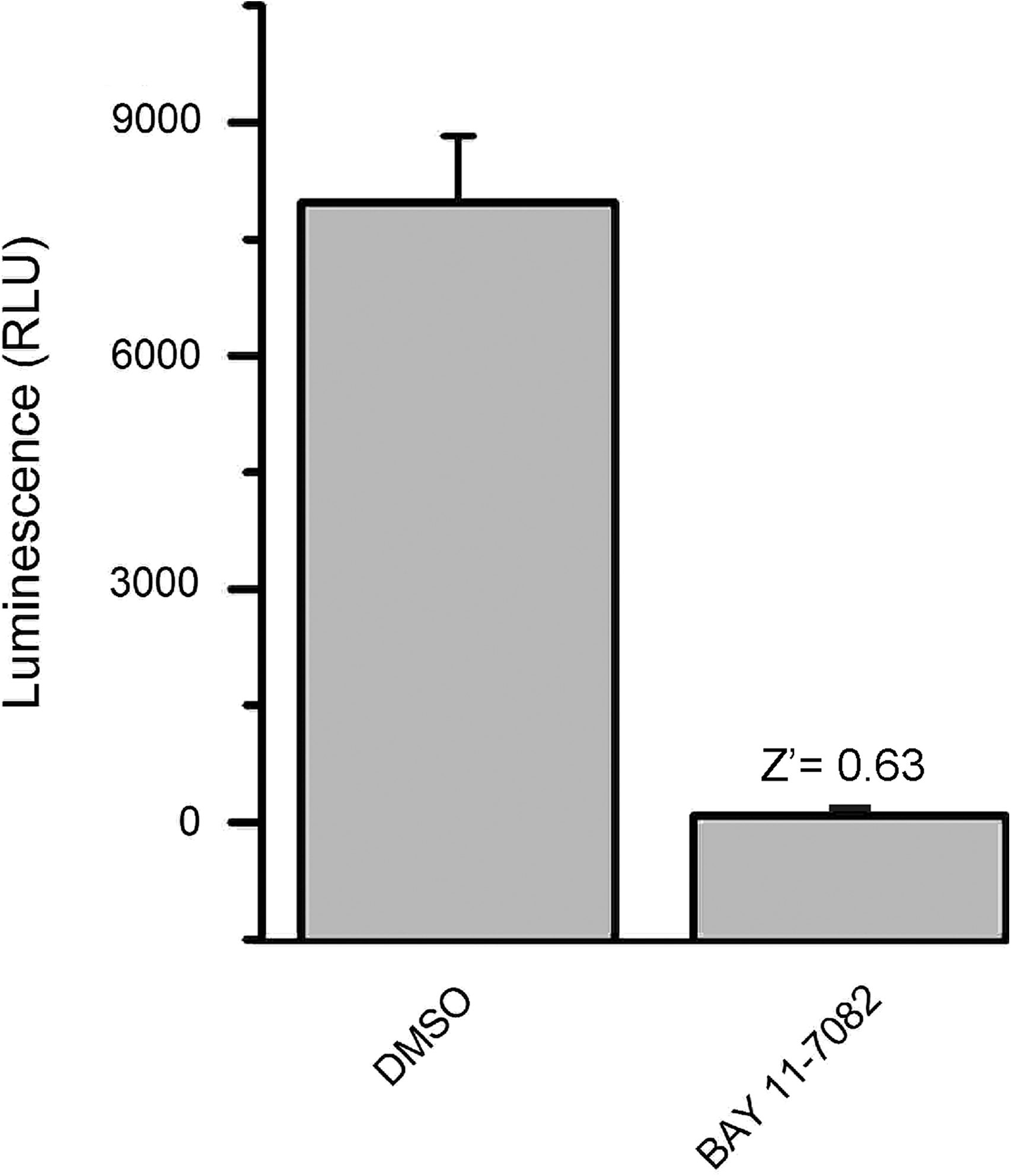
Evaluation of assay reliability. Luminescence values from HEK293 cells transiently transfected with CLuc-FGF14+CD4-Nav1.6-NLuc and either treated with 0.5% DMSO (positive control) or BAY 11-7082 (negative control and/or background) were 7970±150 RLU and 84.2±4.5 RLU, respectively. Signal-to-noise ratio was 142, signal-to-background was 95.3, and signal window was 5.3. Data are mean±SD; the statistical parameters were calculated over n=32 wells for each group.
Discussion
In the studies described here we demonstrate for the first time the use of the LCA as a rapid assay to detect the assembly of the FGF14:Nav1.6 channel C-tail complex, to validate hot spots at this protein–protein interaction complex and to investigate its dynamic regulation by kinases in live cells. We chose to focus on this protein complex for the following reasons. First, neuronal FGF14 is a functionally relevant component of the Nav channelosome that controls Nav channel gating properties and expression with a complexity and potency unique to any other iFGFs or other known Nav interactors. 8 –13 Furthermore, in rodents genetic deletion of fgf14 impairs neuroplasticity and cognitive function, and in humans mutations of FGF14 result in neurodegeneration, 20,21 indicating an important preclinical and translational significance to this brain molecule. 10,12,22,23,56 Of all Nav channel α subunits detected in complex with FGF14, Nav1.6 was chosen because it is the most sensitive to FGF14 modulation. 8 Nav1.6 channels play a critical role in fine tuning neuronal excitability, 57 are linked to human diseases, 58,59 and pharmacological inhibition of Nav1.6 channels is neuroprotective. 60 Thus, the development of this new assay to detect the FGF14:Nav1.6 channel C-tail complex and to rapidly screen its dynamic modulation could provide a launching platform for probe development and drug discovery in the central nervous system.
The bioluminescence-based LCA introduced by Luker et al. 35 provides a quantitative and reversible real-time readout of protein–protein interactions in vitro and in vivo and is an emerging alternative to fluorescence-based assays (for example, FRET 25 ). In our adaptation of this assay, the two complementary N-terminus (NLuc 2-416) and C-terminus (CLuc 398-550) fragments of firefly luciferase were, respectively, fused into a chimera of the CD4 transmembrane segment and the Nav1.6 C-tail (CD4-Nav1.6-NLuc) or human FGF141b isoform (CLuc-FGF14). The use of a construct expressing solely the Nav1.6 C-tail presents several advantages: (i) it is an alternative to the expression of full-length recombinant Nav channels, which are high molecular weight transmembrane proteins hard to express into heterologous expression systems; (ii) it preserves the Nav channel C-tail natural orientation juxtaposed to the plasmamembrane and its membrane targeting; 39 (iii) it isolates the C-tail from the rest of the Nav channel, limiting any potential indirect modulatory effects on the protein–channel complex induced by other intracellular domains of Nav. However, future optimizations might include the development of stable cell lines expressing full-length Nav channel constructs tagged to one of the luciferase fragments (either N- or CLuc) for use in simultaneous LCA and automated-patch clamp or fluorescence-dye platforms. Co-transfection of CD4-Nav1.6-NLuc and CLuc-FGF14 constructs in HEK293 cells resulted in a robust and stable assembly of the protein–channel C-tail complex over time. Z′ factor, signal-to-background, and signal-to-noise values 37,54 were in agreement with previous in-cell luminescence complementation assays. 61,62 Thus, upon miniaturization and up-scaling from 96-well to 1536-well plates format, we envision that this assay will be suitable for large high-throughput screening.
We have explored the use of the LCA for validating structural changes at the FGF14:Nav1.6 channel complex interface. Our results show that residues Y153 and V155 play a critical role in the interaction with Nav1.6, and in agreement with previous studies, 9 we confirm by in silico analysis of the Y153N/V155N mutations that these residues are part of a hot spot at the FGF14:Nav1.6 channel interface. When evaluated in the LCA, mutations of Y153 and V155 into N led to a reduction of the FGF14:Nav1.6 assembly, with no effects on FGF14 expression levels, confirming our hypothesis that these residues are likely to impose structural changes at the protein–complex interface. Evaluation of additional residues identified in silico will be necessary, however, to fully characterize the structure and specificity of the FGF14:Nav1.6 channel interface. The effect of naturally occurring mutations of either FGF14 (FGF14F145S, SCA27 mutant 21 ) or of the Nav channel C-tail on reciprocal interactions are also important and could be evaluated using the LCA. Interestingly, the folding-defective Nav1.1-M1841T epileptogenic mutant can be partially rescued by co-expression with the β1 accessory subunit, raising the possibility of utilizing protein–protein interactions as a base for medication development against channelopathies. 63 –65 Thus, the LCA not only provides a useful tool for rapid screenings of hot spots at relevant protein–channel interfaces, but it might also be useful to evaluate the impact of disease-linked mutations on the assembly of macromolecular complexes.
We have also explored the dynamic regulation of the FGF14:Nav1.6 complex by intracellular signaling pathways by evaluating the effect of selective kinase inhibitors on the FGF14:Nav1.6 complex assembly, seeking compounds that could potentially control neuronal excitability through protein–protein interaction. We first evaluated the effect of MAPKp38 on the assembly of the FGF14:Nav1.6 complex. In the brain MAPKp38 signaling is central for synaptic function and remodeling, 49 and previous studies have shown a role of iFGFs in facilitating the recruitment of MAPKp38δ to the MAPK scaffold IB2/JIP2, 46 a protein involved in axonal transport. 66 The functional significance of the iFGF binding to IB2 is still poorly understood, but one can speculate an implication of the iFGFs for axonal targeting of Nav channels through the IB2/MAPKp38δ complex. Our results show that pretreatment with the p38 inhibitor PD169316, significantly reduces the FGF14:Nav1.6 complex assembly. Interestingly, direct phosphorylation of Nav1.6 by p38 suppresses current density and promotes channel internalization. 67 –69 Thus, activation of p38 might lead to opposite effects on neuronal excitability depending on whether it results from direct phosphorylation of Nav1.6 or increased stability of the FGF14:Nav1.6 complex. Additional studies are needed to evaluate the functional significance of this pathway in vivo.
We next evaluated the effect of BAY 11-7082, an inhibitor of the NFκB pathway, on the FGF14:Nav1.6 complex assembly. There is an emerging interest for the role of the IKK/NFκB pathway in the brain. In the canonical NFκB activation pathway, the IKK complex phosphorylates the inhibitor of NFκB (IκB), IκBα, leading to its degradation and to the activation of nuclear NFκB. 70 The IKK/NFκB has been implicated in neuronal plasticity, 71,72 neuronal survival and neuronal injury. 73 Interestingly, functional components of the IKK/NFκB signaling cascade are expressed at the AIS of neurons together with members of the iFGF family, 48 contributing to axonal growth and formation of the AIS. 74,75 Some of these effects might result from rapid nongenomic activity of NFκB. 72,76 In this study, we show that inhibition of IKK limits the FGF14:Nav1.6 complex assembly. Thus, stimuli that activate the NFκB pathway in vivo might promote the assembly of the Nav channel to its multivalent accessory protein, FGF14, stimulating excitability. How these predicted outcomes correlate with the emerging role of the NFκB pathway in neuronal polarity 74,75 is of great interest and should be investigated. Overall, our findings provide evidence for a dynamic regulation of the FGF14:Nav1.6 channel C-tail complex by two specific kinase pathways, MAPK/p38 and NFκB. A challenging task would be to extend this study to most, if not all, kinases, and more broadly to evaluate the role of phosphorylation-dependent protein–protein interactions in regulating neuronal excitability. One approach to such large-scale studies might be to systematically analyze the effect of kinases inhibitors on all known combinatorial interactions between Nav isoforms and iFGFs or any other relevant accessory proteins through chemical library screenings. Upon up-scaling and miniaturization, the LCA could conceivably provide the basis for a rapid and high-throughput methodology toward this long-term goal.
One problem associated with using luciferase-based assays in screening studies is the number of false positives arising from the direct effect of compounds on the enzymatic activity of luciferase. A growing number of studies have reported such effects. A relevant example is PTC124 (3-[5-(2-fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid), a molecule originally identified in a cell-based firefly luciferase assay as a potential therapeutic agent, 77 which was later discovered to directly suppress the activity of the luciferase reporter. 50,78,79 To validate the effect of PD169316 and BAY 11-1082, we designed a series of counter screenings against the activity of recombinant luciferase, cell viability, and protein expression. To accelerate data validation and triage false positives at early stages, we propose these validation steps to be an integrative platform for any luminescence-based chemical screening.
In summary, we have adapted the LCA to study the assembly of a protein complex at the level of Nav channels. Based on the sensitivity and reliability of this assay, we envision that this study introduces a novel, rapid, and robust methodology for detecting the dynamic regulation of protein–protein interactions within ion channel complexes real-time in live cells with potential applications in the proteomic and drug discovery field. The large network of protein–protein interactions that composes the macromolecular complex around ion channels and controls neuronal function provides a rich source of drugable interfaces. 80,81 The specificity of these interactions is regulated by hot spots, and peptides, peptidomimetics, and small molecules targeting these hot spots can serve as highly selective and potent scaffolds for drug development. 5,82 The LCA is a rapid and effective method that could be employed for compound screening identification, hit validation, and hit-to-lead transition toward drug discovery development against Nav channels and other challenging transmembrane ion channels. 83 The new generation of the split-luciferase reporters based on the heterocomplementation of green and red luciferase 84 extends the capability of the assay to detect three-way combinations of interacting partners, providing even broader possibilities for studying the ion channel interactome.
Footnotes
Acknowledgments
The authors thank Dr. Sarkar Partha (Department of Neurology, UTMB) for providing the firefly full-length luciferase; Dr. David Piwinca-Worms (Washington University, St. Louis, MO) for the gift of the plasmids expressing FRB-NLuc and FKBP-CLuc; Dr. Benedict Dargent (INSERM, France) for the CD4-Nav1.6 construct; Dr. Van Swieten (Department of Neurology, Erasmus Medical Center, Rotterdam, The Netherlands) for providing the PDB coordinates of the FGF14 molecular model; and Marcy Bubar Jordan (Center for Addiction Research, UTMB) for proofreading the manuscript. This work was supported by a grant from the Institute for Translational Sciences from UTMB (F.L.), a Research Starter grant from the PhRMA Foundation (F.L., S.S.M.), the American Heart Association (S.S.M.), and 5UC7AI07008304 (R.V. and N.B.).
Disclosure Statement
No competing financial interests exist.