
Abstract
In eukaryotes, the spindle checkpoint acts as a surveillance mechanism that ensures faithful chromosome segregation. The spindle checkpoint prevents premature separation of sister chromatids and the onset of anaphase until every chromosome is properly attached to the mitotic spindle. Tumorigenesis might result from generation of aneuploidy by dysfunction of the spindle checkpoint. Differences of the checkpoint system in normal cells versus tumor cells might provide a new opportunity in cancer drug development; therefore, efforts to identify the spindle checkpoint inhibitors have been fostered. Based on spindle checkpoint inhibitors being able to induce cells to exit mitotic arrest caused by microtubule drug treatment, we developed a cell-based assay to screen compounds that were potential spindle checkpoint inhibitors. This assay was validated with a known spindle checkpoint inhibitor and was easy to adapt to a large-scale screening. It also had the advantages of being high in sensitivity and low in cost.
Introduction
Malfunction of the spindle checkpoint can lead to chromosome mis-segregation and premature exit from mitosis, which results in aneuploidy, and even tumorigenesis. 1 Partial inactivation of the spindle checkpoint has been found in many tumor cell lines, including ovarian, colon, hepatocellular carcinoma, breast cancer, lung cancer, and so on, 5 –8 and impaired mitotic checkpoint has been implicated as a contributor to tumorigenesis. 9,10 In addition, it was reported that mice heterozygous for spindle checkpoint genes had a tendency to develop tumors at high rates after long latencies. 11 However, the spindle checkpoint is essential for cell viability because homozygous knockout mice lacking the functional spindle checkpoint fail to survive. 12,13 Inactivation of the spindle checkpoint by RNAi induces mitotic defects that are lethal to cells. 14 Moreover, it has been reported that compared with normal cells, tumor cells display higher sensitivity to the spindle checkpoint inhibitor. 15 Taking these findings into account, the differences of the spindle checkpoint system between normal and tumor cells make the spindle checkpoint a novel target for anticancer therapy.
To identify inhibitors of the spindle checkpoint, a high-throughput screening assay is necessary. Here, we described an efficient cell-based assay for screening chemical inhibitors of the spindle checkpoint. This high-throughput assay is easy to perform and does not require special instruments. In addition, the assay has advantages of being sensitive and low cost.
Materials and Methods
Cell Culture and Cell Lines
HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM; GIBCO), supplemented with 10% (v/v) fetal bovine serum (GIBCO), 100 units/mL penicillin and 100 μg/mL streptomycin, and were maintained at 37°C in a humidified atmosphere of 5% CO2.
Reagents and Antibodies
Compound 12W was obtained from Alputon Inc. Nocodazole, MG132, and 4′,6-diamidino-2-phenylindole (DAPI) were supplied by Sigma-Aldrich Co. These reagents were dissolved in dimethylsulfoxide (DMSO) as a stock solution and stored at −20°C. The rabbit polyclonal antibody against phospho-histone H3 (Ser-10) (P-H3) was purchased from Cell Signaling Technology. The mouse monoclonal antibodies against Aurora A and Aurora B were from BD Transduction, and the mouse monoclonal antibody against β-actin was purchased from Sigma-Aldrich. Goat anti-rabbit and goat anti-mouse immunoglobulin G (IgG) horseradish peroxidase conjugates were bought from Pierce Biotechnology, Inc.
MTT assay
After mitotic cells were washed away from the 96-well plate, DMEM containing 0.5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added and the plate was incubated for an additional 2–4 h in a CO2 incubator at 37°C. The medium was then discarded and DMSO was added to dissolve the formazan crystals, which turned the solution purple. The absorbance at 570 nm was measured using a plate reader (Molecular Devices).
Western Blot Analysis
After drug treatment, cells were collected by centrifugation. Whole cell extracts were obtained by lysing cells in a buffer containing 20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM ethylene diamine tetraacetic acid, 1% Triton-X 100, 1 mM phenylmethanesulfonyl fluoride, 10 μg/mL leupeptin, 2 μg/mL aprotinin, 10 mM NaF, and 1 mM Na3VO4. After incubation for 30 min on ice, the protein lysate was cleared of debris by centrifugation at 10,000 g for 5 min. Supernatant was collected and protein concentration was determined by the bicinchoninic acid assay (Pierce Biotechnology, Inc.). Twenty to 50 μg of proteins was separated by 12%–15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to the polyvinylidene diflouride membrane (Millipore). Following incubation with TBS-Tween 20 (0.05%; TBS-T) and 5% nonfat milk for 2 h at room temperature, the membrane was incubated in fresh blocking solution with an appropriate dilution of primary antibody for 2 h at room temperature. The membrane was washed for 5 min in TBS-T three times and then incubated with horseradish peroxidase–conjugated goat anti-rabbit IgG or goat anti-mouse IgG for 1 h at room temperature. Membrane was again washed for 5 min in TBS-T three times and then developed by enhanced chemiluminescence (Pierce Biotechnology, Inc.). Protein expression levels were determined by densitometry with Quantity One software (Bio-Rad), normalized to the level of β-actin.
Cell Cycle Analysis
The effect of nocodazole on cell cycle progression was analyzed by flow cytometry. After drug treatment, cells were collected by centrifugation, washed twice with phosphate-buffered saline (PBS), and fixed with 70% (v/v) ethanol at 4°C over night. They were then treated with 100 μg/mL RNase A (Sigma-Aldrich), and incubated at 37°C for 30 min before staining with 100 μg/mL propidium iodide (Sigma-Aldrich) overnight at 4°C. Cell cycle analysis was carried out using the Coulter EPICS XL flow cytometer (Beckman Coulter Inc.,).
Mitotic Index
HeLa cells were plated in six-well plates and treated with nocodazole alone or followed by 30 μM 12W treatment for 2 h. Medium was aspirated, and cells were then washed with PBS, fixed with 3.7% formaldehyde for 10 min at room temperature, and incubated at room temperature with the staining solution (0.3% Triton-X 100, 100 μg/mL RNase A and DAPI in PBS) for 5 min avoiding light. For each sample, 100–500 cells were randomly counted by fluorescence microscopy, and mitotic cells were scored, with a lack of nuclear membrane in cells indicating chromosome condensation.
Immunofluorescence Staining
HeLa cells grown on 22-mm2 cover slips in six-well plates were washed once with PBS and fixed with 3.7% formaldehyde in PBS for 10 min at room temperature. Cells were then rinsed with PBS before treated with a washing buffer (PBS containing 0.2% Triton X-100, 0.2% bovine serum albumin [BSA], and 1 mM NaN3) on ice for 10 min. The cover slips were then incubated with the blocking buffer (PBS buffer containing 0.02% Triton X-100, 3% BSA, and 1 mM NaN3) for 30 min at room temperature. Cells were then incubated with 2 μg/mL anti-P-H3 in the blocking solution for 30 min. Next, cells were washed four times with a washing buffer (PBS containing 0.02% Triton X-100, 1.5% BSA, and 1 mM NaN3), followed by incubation with 20 μg/mL of goat anti-rabbit Alexa Fluor 647 (Invitrogen) in the blocking solution for 30 min. Cells were washed four times with the washing buffer (PBS with 0.02% Triton X-100, 1.5% BSA, and 1 mM NaN3) before incubation with DAPI (1 μg/mL) in washing solution for 2 min. Specimens were mounted in 90% glycerol and sealed with nail polish.
Statistical Analysis
To determine significant differences in the data, the Student's t test was applied. Results were considered to be significantly different at P<0.05. Pearson's correlation coefficient (r) was used to measure the correlations between mitotic index and optical density at 570 nm (OD570) at the same concentration of 12W. The dose–response data were subjected to nonlinear regression analysis using the GraphPad Prism software.
Results
A High-Throughput Assay for Screening Inhibitors of the Spindle Checkpoint
A high-throughput assay was established to screen inhibitors of the spindle checkpoint based on characteristics of cells at different stages. During interphase, adhesion cells were tightly attached to the supporting matrix in the culture dishes (Fig. 1). During mitosis, the cells became rounded and detached from the supporting matrix. After completing mitosis, cells became flattened and re-attached to the cell culture dish. Microtubule inhibitors such as nocodazole can trigger the spindle checkpoint and arrest cells in mitosis, and such rounded cells can be easily collected by shaking the cell culture dish gently. Cells arrested in mitosis were transferred to 96-well plates and incubated with the test compounds. Compounds that could inhibit the spindle checkpoint would induce cells to exit mitosis, flatten, and re-attach to the supporting matrix. Negative compounds would not be able to induce similar effects, and cells treated with them would remain rounded and could be easily washed from cell culture dishes. We used the MTT assay to measure the relative number of remaining cells. MTT was reduced from the original yellow solution to purple formazan crystal by succinic dehydrogenase in the mitochondria of living cells. When DMSO was added to dissolve the formazan crystals, a purple solution formed and absorbance at 570 nm was measured with an enzyme-linked immunosorbent assay microplate reader; the OD570 reading was proportionate to the number of cells. A large amount of remaining cells suggested the compound might be a spindle checkpoint inhibitor. Based on the absorbance reading, we could determine which compounds led to mitotic exit in nocodazole-treated cells.
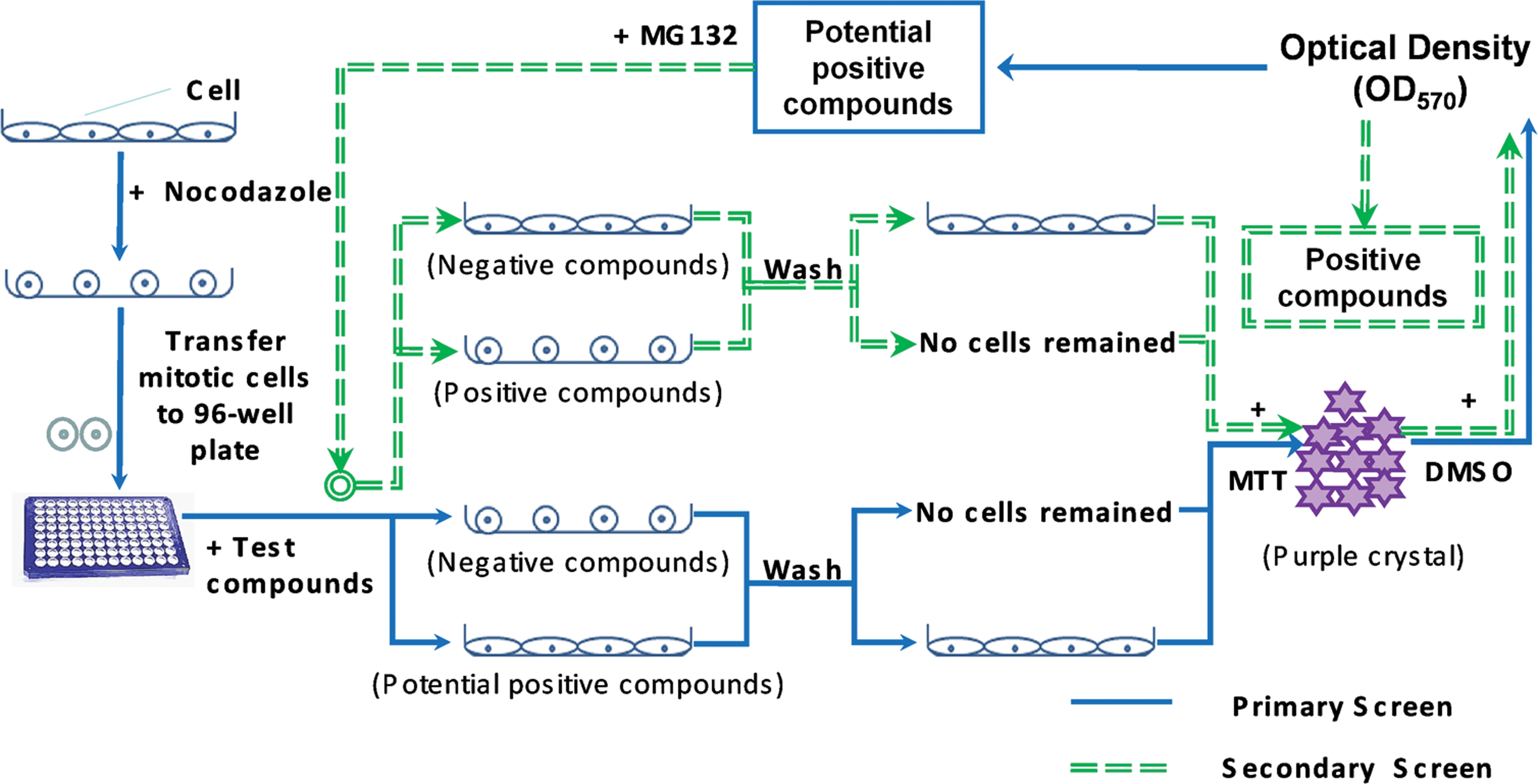
Schematic of a cell-based assay for screening the spindle checkpoint inhibitors. In response to microtubule drugs such as nocodazole, adhesion cells (HeLa cells) were arrested in mitosis and became rounded. They were forced to exit mitosis and regain adhesion ability when treated with the spindle checkpoint inhibitors. Without the spindle checkpoint inhibitors, mitotic cells remain rounded and could be washed away from the plate. Afterward, the MTT assay was used to measure the number of cells remaining on the plate, which was reflected by the optical density reading at 570 nm. Potential inhibitors could be selected from the primary screen as indicated by solid arrows. A secondary screen was designed as indicated by dashed arrows to eliminate false positives from potential inhibitors. In the secondary screen, nocodazole-arrested cells were treated with potential spindle checkpoint inhibitors selected from the primary screen and a proteasome inhibitor MG132. The spindle checkpoint inhibitors could induce mitotic exit among nocodazole-arrested cells in the absence of proteasome inhibitors but could not induce mitotic exit in the presence of proteasome inhibitors. MTT, MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; DMSO, dimethylsulfoxide; OD570, optical density at 570 nm. Color images available online at
The spindle checkpoint negatively regulates the ubiquitin–proteasome pathway and prevents cyclin B and other proteins from degradation by the proteasome. Therefore, the proteasome activity is absolutely necessary for the spindle checkpoint inhibitors to induce mitotic exit. Inhibition of the ubiquitin–proteasome pathway would result in failure of the spindle checkpoint inhibitors to induce mitotic exit. Based on this assumption, we designed a secondary screen (Fig. 1) to exclude false-positive compounds from those selected by the primary screen. False-positive compounds could induce mitotic exit through other mechanisms, such as inhibition of cyclin-dependent kinase (CDK). In the secondary screen, nocodazole-arrested cells were treated with the proteasome inhibitor MG132 and the potential positive compounds selected from the primary screen, as seen in Figure 1. Compounds that can induce nocodazole-arrested cells to exit from mitosis in the absence of MG132 but are unable to do so in the presence of MG132 would be considered positive inhibitors of the spindle checkpoint.
12W, an Aurora Inhibitor, Can Induce Mitotic Exit
We chose 12W, a recently reported Aurora kinase inhibitor, 16 to test the feasibility of our high-throughput system. First of all, we analyzed whether 12W can inhibit Aurora kinase activity in a cell-based assay. Histone-H3 is considered an Aurora kinase substrate, and the level of histone-H3 phosphorylated at Serine 10 (P-H3) will decrease when Aurora kinase activity is inhibited. 17 We treated nocodazole-arrested HeLa cells with different concentrations of 12W and then measured the expression of P-H3, Aurora A, and Aurora B by Western blot. As shown in Figure 2A, treatment of HeLa cells with 12W resulted in decreasing the level of P-H3, Aurora A, and Aurora B in a dose-dependent manner. Immunofluorescence results also indicated that 12W could reduce intracellular P-H3 (Fig. 2B). In agreement with previous reports, all our results suggest that 12W is an inhibitor of Aurora kinase. We then analyzed whether inhibition of Aurora kinase could abolish the spindle checkpoint since Aurora kinase was implicated in the spindle checkpoint function. 18 –20 The nuclear membrane of cells that were treated with nocodazole disappeared and chromosomes condensed (Fig. 3A). However, in the group of further treatment with 12W, chromosome decondensation was observed, indicating that 12W induced mitotic exit in the presence of nocodazole. Meanwhile, we found that some nuclei that had re-appeared had abnormal shape (Fig. 3A). Furthermore, mitotic index was calculated based on the percentage of cells with condensed chromosomes. We found that 12W caused the mitotic index to decrease from 80% to 10% in a dose-dependent manner, with an IC50 value of 10.92 μM (Fig. 3B). However, mitotic exit induced by 12W could be blocked by the proteasome inhibitor MG132 (Fig. 3C), which indicated that 12W relieved the inhibition of the APC/C caused by the spindle checkpoint. These results demonstrated that 12W could inhibit the mitotic spindle checkpoint activated by microtubule inhibitors such as nocodazole.

Compound 12W could inhibit phospho-histone H3 (Ser-10) (P-H3) and down-regulate expression of Aurora A and Aurora B.

Compound 12W can override mitosis arrest induced by nocodazole.
Evaluation of the Assay for Screening Inhibitors of the Spindle Checkpoint
Having shown that 12W is an inhibitor of the spindle checkpoint, we used 12W as a positive control to validate our high-throughput assay. Nocodazole-arrested HeLa cells were collected and transferred to 96-well plates. Then 12W was added in the presence or absence of the proteasome inhibitor MG132. After incubation for 4 h, cell culture medium was discarded from each well, and the medium containing MTT was then added. OD570 was determined with conventional MTT assay after 2–4 h. As shown in Figure 4A, the abscissa denotes the concentration of 12W and the ordinate denotes the OD570. Increasing OD570 reading was in accordance with increasing concentration of 12W in the absence of MG132 (Fig. 4A), suggesting that more cells overrode mitosis arrest accompanying with increasing 12W and consequently led to an increase in the number of attached living cells. This result inversely correlated with the result presented in Figure 3B showing that 12W decreased the mitotic index of nocodazole-arrested cells in a concentration-dependent manner. We found that 30 μM of 12W could significantly inhibit the spindle checkpoint (Fig. 3B), and the OD570 of cells treated with 30 μM of 12W was six times larger than that of untreated cells detected by this high-throughput assay. We then analyzed the correlation between the OD570 value and the mitotic index corresponding to the same concentration of 12W by applying Pearson's correlation coefficient (r). Pearson's correlation coefficient measures the degree to which two variables are linearly related. The closer the absolute value of r is to 1, the better correlation between the two variables. As shown in Figure 4B, the correlation coefficient between OD570 value and mitotic index was −0.982, which indicates the two were highly correlated. However, when HeLa cells arrested in mitosis by nocodazole were treated with increasing concentrations of 12W and 10 μM MG132, similar results were not obtained. The reading of OD570 did not show any significant difference between the control group and the experimental group (Fig. 4A). This result suggested that 12W failed to induce mitotic exit among nocodazole-arrested cells in the presence of MG132. Such a result is in accordance with the result depicted in Figure 3C that 12W could not reduce the mitotic index when MG132 was added. In conclusion, the final absorbance value of our screening assay could well reflect activity of the spindle checkpoint inhibitors.
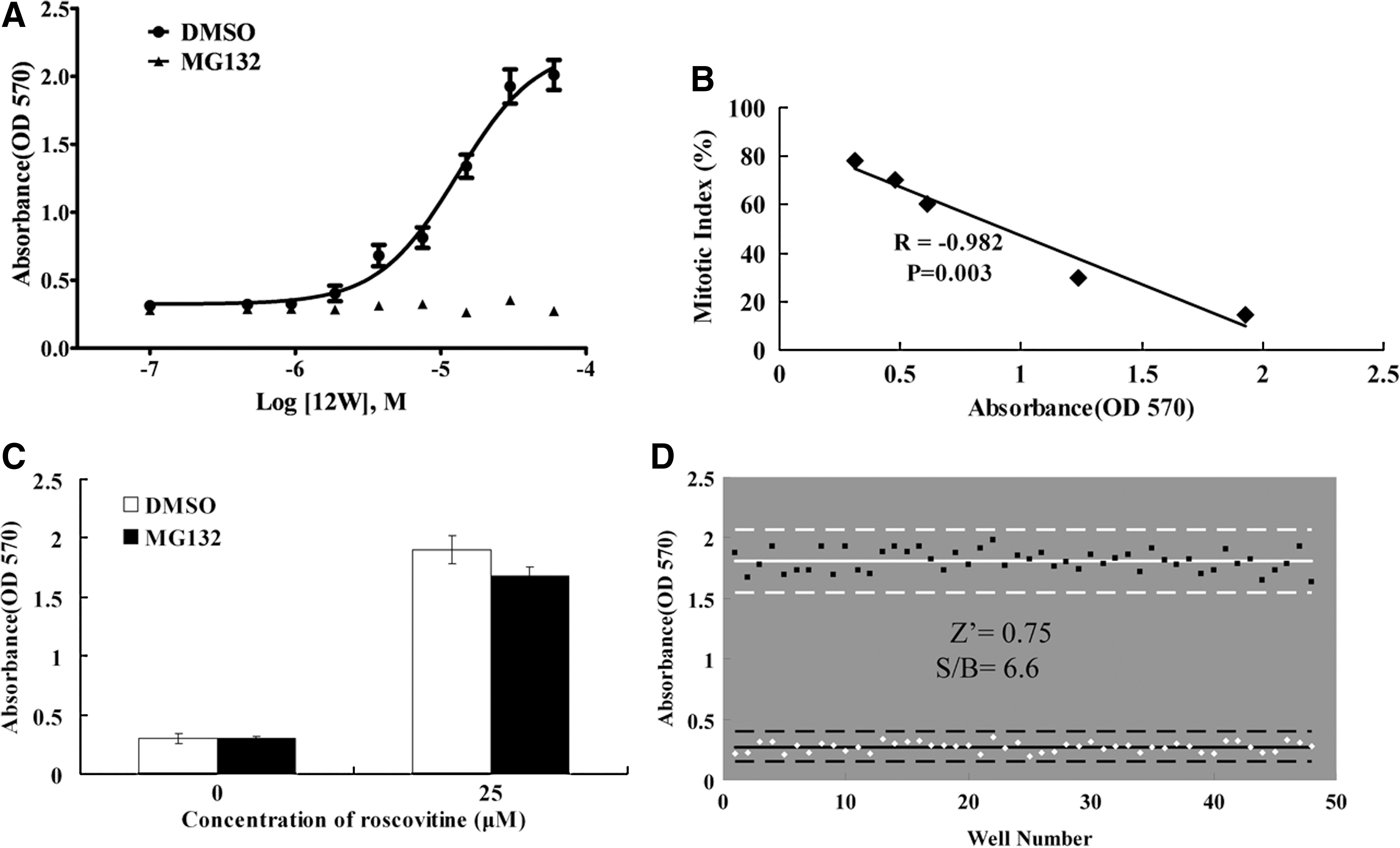
The assay was validated by correlation coefficient and Z′ factor analysis.
A CDK inhibitor, roscovitine, was used to validate the secondary screening. Compared with the control group, treatment of mitotic cells with roscovitine led to a significantly increased OD570 both in the absence and presence of MG132 (Fig. 4C), so false-positive compounds that induce mitotic exit through other mechanisms such as inhibition of CDK could be excluded from compounds selected from the primary screen.
The Z′ factor provides a statistical measurement for the quality of a screening assay, with a Z′ factor between 0.5 and 1.0 considered suitable for high-throughput screening. 21 To assess the reproducibility of this assay, we compared 30 μM 12W treatment to DMSO vehicle control treatment across an entire 96-well plate as shown in Figure 4D. The Z′ factor of 0.75 was obtained with data we collected, and the signal to background value was 6.6. These findings demonstrated that the assay is statistically suitable and robust for small molecule screening applications.
Optimization of the High-Throughput Assay to Screen the Spindle Checkpoint Inhibitors
To optimize the conditions for inducing mitotic arrest at M phase by nocodazole treatment at the beginning of drug screening, we used flow cytometry to examine the effect of nocodazole on mitotic arrest. Nocodazole-induced cells arrested in G2/M in a time- and concentration-dependent manner (Fig. 5A and B). Most of the cells were arrested in the G2/M phase after being treated with 100 ng/mL nocodazole for 16 h compared with the control group. In addition, it is known that the level of P-H3 can serve as a marker of mitosis because P-H3 appears to occur exclusively during mitosis in mammalian cells, histone H3 dephosphorylation occurs quite rapidly when cells exit from mitosis, and Ser-10 remains unphosphorylated throughout the interphase. After HeLa cells were treated with 100 ng/mL nocodazole for different time intervals, we measured the level of P-H3 by Western blot (Fig. 6A). We found that nocodazole treatment led to increased expression of P-H3 in a time-dependent manner; however, we noticed that cells underwent apoptosis when treated with nocodazole for more than 20 h (data not shown). We measured the level of P-H3 in HeLa cells treated with various concentrations of nocodazole for 16 h, and we found that it was upregulated when the concentration of nocodazole was 100 ng/mL (Fig. 6B). However, no further increase in the level of P-H3 was observed in cells treated with 200 ng/mL nocodazole. These results suggest that nocodazole treatment at a concentration of 100 ng/mL for 16 h would lead to the majority of cells being arrested in mitosis.

The effect of nocodazole on the cell cycle. The cell cycle was analyzed by flow cytometry after being treated with nocodazole. HeLa cells were treated with
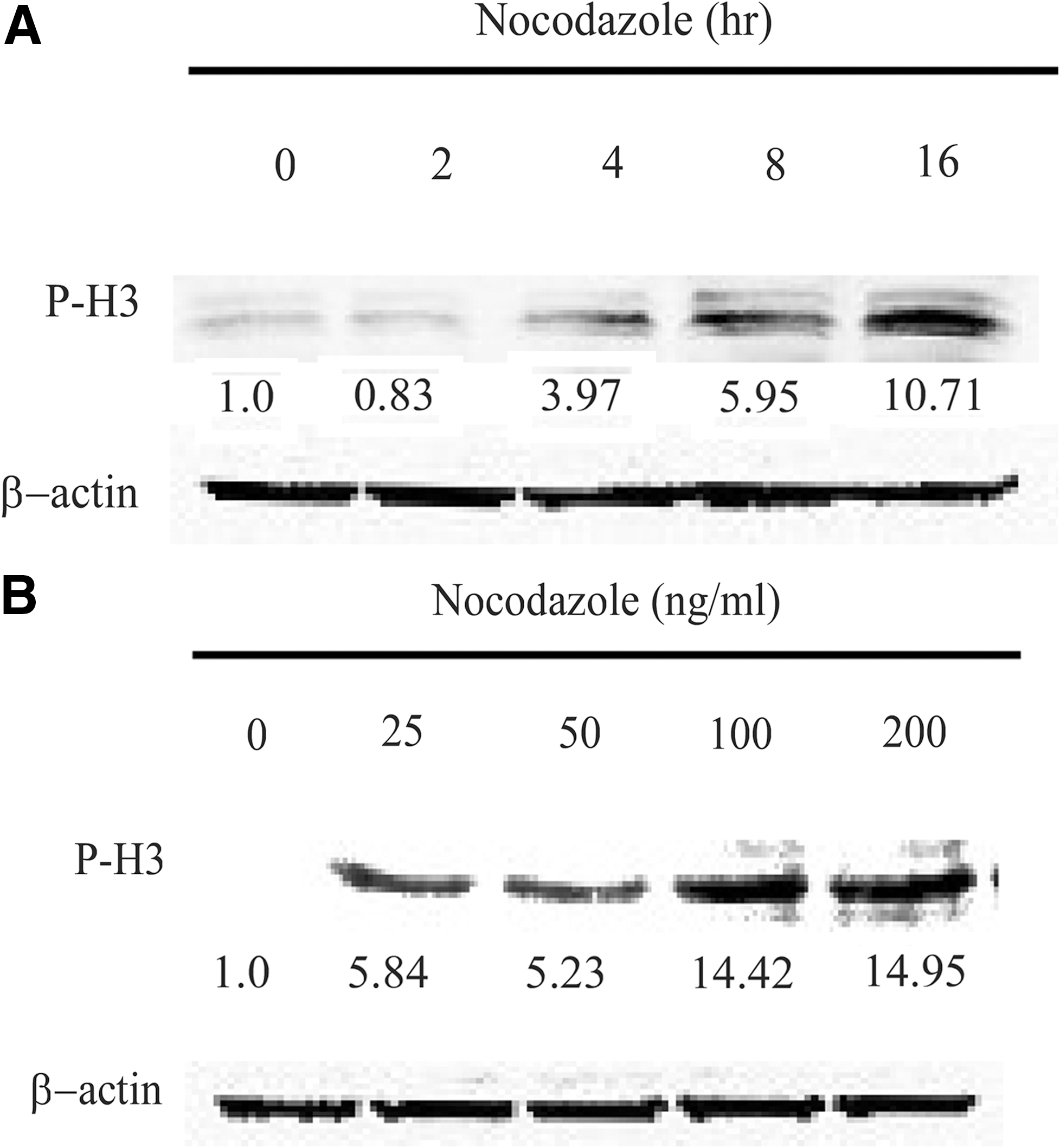
P-H3 was increased dramatically due to nocodazole treatment. HeLa cells were treated with
After HeLa cells were arrested in mitosis, we transferred mitotic cells into 96-well plates and optimized the initial seeding number of cells. Cells were transferred to 96-well plates at a density of 5,000, 10,000, and 20,000 cells per well, respectively, and 12W was added at different concentrations, followed by measurement of OD570 in each well. As shown in Figure 7A, corresponding to the same concentration of 12W, the OD570 was better correlated with the mitotic index (Fig. 3B) when the initial cell count was 20,000 per well. The correlation coefficient of each group (Fig. 7B) also reflects that the group containing 20,000 cells per well at the beginning was superior to the other groups. In summary, the following procedure as outlined in Table 1 is recommended for screening the spindle checkpoint inhibitors. HeLa cells were first cultured in 10-cm culture dishes before being treated with 100 ng/mL nocodazole for 16 h, and mitosis-arrested cells were collected by shaking the dishes gently. Then, 90 μL of cell suspension containing 20,000 cells was transferred to each well of a 96-well plate, followed by addition of 10 μL of test compounds (compound library stored in 96-well plates can be diluted to proper drug concentration accordingly), while nocodazole was maintained at 50 ng/mL. The plates were incubated in a 37°C incubator for 4 h before being washed with PBS once, and 100 μL of culture medium that contained 0.5 mg/mL MTT was added to each well of the plates, which were then incubated again for 2–4 h in the dark, after which the supernatant was discarded. After 100 μL of DMSO was added, plates were gently shaken on a plate shaker for 10 min, then the OD570 was measured with a microplate reader. The secondary screening was implemented after the primary screening was complete. Basically, the secondary screening was similar to the primary screening except potential positive compounds selected from the primary screening and 10 μM MG132 were incubated with the mitosis-arrested cells instead of the compound library used in the primary screen. Cells treated with DMSO were used as negative controls, and those treated with 30 μM of 12W were positive controls. The compounds that induced a significant increase in OD570 in the primary screening but failed to do so in the secondary screening with presence of a proteasome inhibitor MG132 were considered as positive inhibitors of the spindle checkpoint, and could undergo further analysis.

Optimization of number of nocodazole-arrested cells seeded into 96-well plate. ) cells per well, followed by incubation with an increasing concentration of 12W for 4 h, and then OD570 was determined as described in Figure 4A. Values represent the means from three separate experiments performed in triplicate±SEM.
Protocol for the Cell-Based Assay for Screening Inhibitors of Spindle Checkpoint
1. HeLa cells cultured in 10-cm dishes and maintained at 37°C in a humidified atmosphere of 5% CO2.
2. Nocodazole added to treat HeLa cells cultured in 10-cm dishes.
3. Cells arrested in mitosis and become rounded.
4. Mitotic cells collected by shaking the dish gently.
5. Compound library stored in 96-well plates; potential positive compounds selected from the primary screening.
6. MG132 added with potential positive compounds in the secondary screening.
7. Cells incubated.
8. Cells washed gently.
9. 100 μL/well solution added in dark.
10. Cells incubated in dark.
11. Plates gently shaken on a plate shaker for 10 min.
12. Cells treated with DMSO were negative controls, and those treated with 30 μM of 12W were positive controls.
DMEM, Dulbecco's modified Eagle's medium; DMSO, dimethylsulfoxide; FBS, fetal bovine serum; PBS, phosphate-buffered saline; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
Discussion
The essential nature of the spindle checkpoint makes it an interesting target for developing anticancer drugs. 14,22,23 Here we reported a high-throughput assay to screen inhibitors of the spindle checkpoint. 12W compound was used as a positive control to test the assay, and final absorbance at 570 nm was in accordance with inhibition of the spindle checkpoint by the compound.
Cells that exited mitosis could be distinguished from cells arrested in mitosis based on their morphological shape and ability to adhere to the supporting matrix. Positive spindle checkpoint inhibitors would induce mitotic exit in nocodazole-arrested mitotic cells, while negative ones would not. In the following washing step, mitosis-arrested cells could be discarded due to their weak adherence. However, the cells able to exit mitosis would not be washed away since they could re-attach to the bottom of the culture plate. Based on this feature, the number of remaining cells was measured by MTT assay, which is usually performed in large-scale screening of anticancer drugs and cytotoxicity tests. The final reading of the MTT assay reflects the number of living cells. The MTT assay is known as a simple, sensitive, and economical method to determine the relative number of cells. Furthermore, the final reading can be readily measured with a microplate reader without other special instruments. Combining the MTT assay with mitosis-arrested cells induced by nocodazole, we established a cell-based assay for a high-throughput system to screen inhibitors of the spindle checkpoint. This system possessed many favorable factors, such as its practicability and sensitivity, in addition to low background interference.
Various mitotic proteins are involved in the spindle checkpoint, and it is accordingly necessary to further identify final targets of the spindle checkpoint inhibitors in order to evaluate the potential of compounds in cancer treatment. DeMoe et al. 23 and Stolz et al. 15 undertook experiments on screening the spindle checkpoint inhibitors and found inhibitors could remarkably inhibit Aurora kinase family member, like Aurora A and Aurora B. Aurora B is a part of the spindle checkpoint system, 18 –20 which may explain inhibition of the spindle checkpoint by these inhibitors. Aurora kinases are over-expressed in a variety of tumor cells, which suggests the Aurora kinase family plays an important role in carcinogenesis; therefore, these family members could represent appealing drug targets for cancer therapy. 24,25
In conclusion, we established a cell-based high-throughput assay to screen inhibitors of the spindle checkpoint; this assay was validated with a positive inhibitor of the spindle checkpoint and was optimized with several crucial parameters. It can be used to screen the spindle checkpoint inhibitors in a high-throughput format with large compound libraries in future. Our work in discovering potential spindle checkpoint inhibitors with this assay is on-going.
Footnotes
Acknowledgments
This work was supported by a grant from Fujian Province Natural Science Foundation of China (2009J01197) and funding from Xiamen Technology Innovation Foundation of China (3502Z20100087). The compound 12W was kindly provided by Alputon Inc. in China.
Disclosure Statement
No competing financial interests exist.