
Abstract


Dr. Robert Lowery received his undergraduate degree in biochemistry from the University of California, Berkeley and his PhD in biochemistry from University of Wisconsin-Madison. Prior to starting BellBrook Labs, Dr. Lowery served for 10 years as Vice President of R&D at PanVera Corporation and previously served as Director of Manufacturing at Promega Corporation. At PanVera, Dr. Lowery played a key role in bringing to market the first fluorescence-based high-throughput screening (HTS) assays for nuclear receptors and protein kinases. At BellBrook, Dr. Lowery has led the development and commercialization of the Transcreener® HTS Assay platform and the iuvo™ Microconduit Array plates. Dr. Lowery's current studies are focused on the identification of selective inhibitors for regulators of G protein signaling (RGS) proteins and the development of biochemical and cellular assays to target neurodegenerative disease pathways.
Dr. Lowery, what was the motivation behind your founding of BellBrook Labs? How did you get the company started, and what were its initial goals and activities? How would you describe BellBrook Labs' current focus and main technologies?
I have been involved in developing screening assays as tools for drug discovery since the early 1990s. I started at a company called PanVera, producing recombinant proteins for HTS for Pfizer and a number of other companies, and then we started developing some of the first fluorescent assays for kinases, nuclear receptors, and the like. By the time BellBrook was started in 2002 there was a mindset that screening as many targets against as many compounds as possible was the way to go. Clinical attrition was acknowledged as a big problem, and the way to overcome it was seen as ramming more things through faster, essentially top-loading the pipeline.
But it was already becoming clear that this would not be the most effective approach. By trying to squeeze more into the HTS infrastructure, the biology was being force-fit or ignored to a significant degree. I wanted to develop a set of tools that would provide access to physiologically relevant in vitro assays—both cellular and biochemical assays—and to a broader swath of human biology. That was my motivation for starting BellBrook.
We initially funded the company with a Small Business Innovation Research (SBIR) grant that I had been awarded while I was working at PanVera. The grant was focused on developing an assay for an obscure class of enzymes called glycosyltransferases. That is how the Transcreener® technology was discovered. What we initially tried did not work, so we came up with this crazy idea of using an antibody to detect a nucleotide product, UDP. All the literature said that if you injected an animal with UDP you would get antibodies to UMP, which seemed logical. But we were desperate at the time, so we tried it, and lo and behold we got antibodies to UDP. The obvious transition to kinases, a much bigger market, led to the development of the ADP assay. Since then we have funded the company with a combination of SBIR grants and private investor financing.
You have been very successful at developing assays based on antibody recognition of nucleotides. Do you think this strategy can be applied to the detection of a much wider range of enzyme products and cofactors? Are there particular classes of such molecules that are difficult to detect using antibodies?
Antibody-based detection does not make much sense for receptors, proteases, or oxidoreductases. But immunoassays are used for just about every other type of enzyme, because antibodies are still the best reagents for selective recognition of enzyme products versus substrates, especially for enzymes that catalyze post-translational modifications, many of which, like kinases or methyltransferases, are drug targets because they play a regulatory role. Immunoassays are the dominant type of enzyme assays used, either for detecting the modified product, such as a phosphorylated peptide or protein, or for detecting the nucleotide product.
Transcreener assays rely on homogenous immunodetection of nucleotide enzyme products with fluorescent readouts (fluorescence polarization, time-resolved fluorescence–fluorescence resonance energy transfer [TR-FRET], and fluorescence intensity). Four different assays enable detection of thousands of enzymes from diverse families.
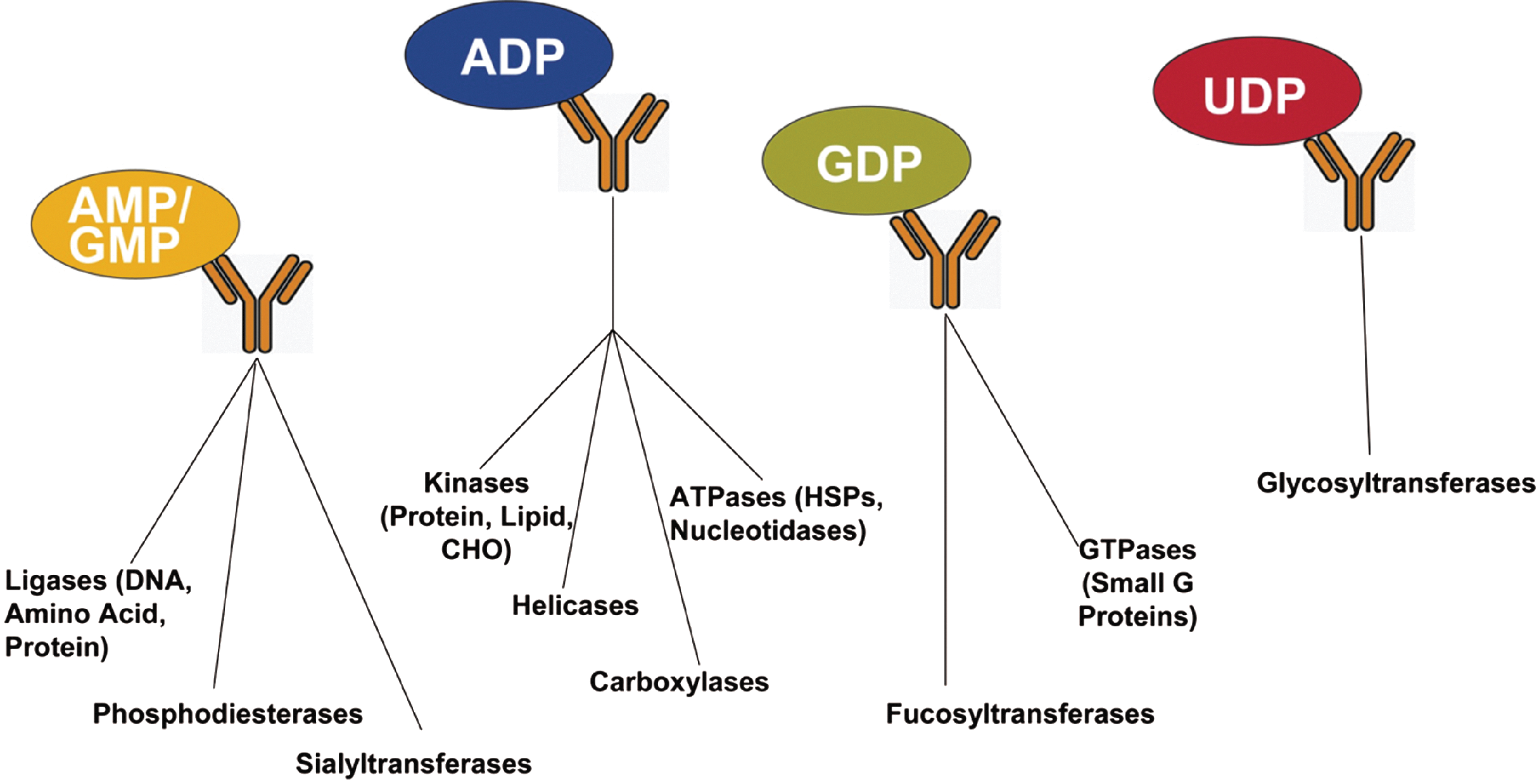
The Transcreener biochemical assay technology is available in either a fluorescence polarization (FP)-based or time-resolved fluorescence–fluorescence resonance energy transfer (TR-FRET)-based format. How do these two detection formats complement each other? What types of applications warrant the use of one versus the other?
It really depends on people's preferences; that's why we have the two formats. FP is simpler, with fewer moving parts, so generally when we develop a new assay we use FP first. The choice also depends on the instrumentation available in a particular laboratory, the types of compounds in the libraries being screened, and the user's comfort level with different types of assays and screens. Both FP and TR-FRET—and some of our assays that are also available in fluorescence intensity (FI) format—are very robust, tried-and-true HTS methods. They have ratiometric readouts, use far red fluors, and are not very susceptible to compound inhibition. FP has a more stable signal than TR-FRET, but with TR-FRET you have a delay in the signal, which may decrease fluorescence interference. Ultimately, though, the makeup of a given library will dictate whether FP or TR-FRET will give less compound interference.
Regarding specific applications, if someone is doing a large screen, say a million compounds or more, I generally recommend using FP because it yields such a stable assay signal. Large screens typically have longer periods between assay and read times. The signal in an FP assay may be stable for days, and this is generally not the case for TR-FRET.
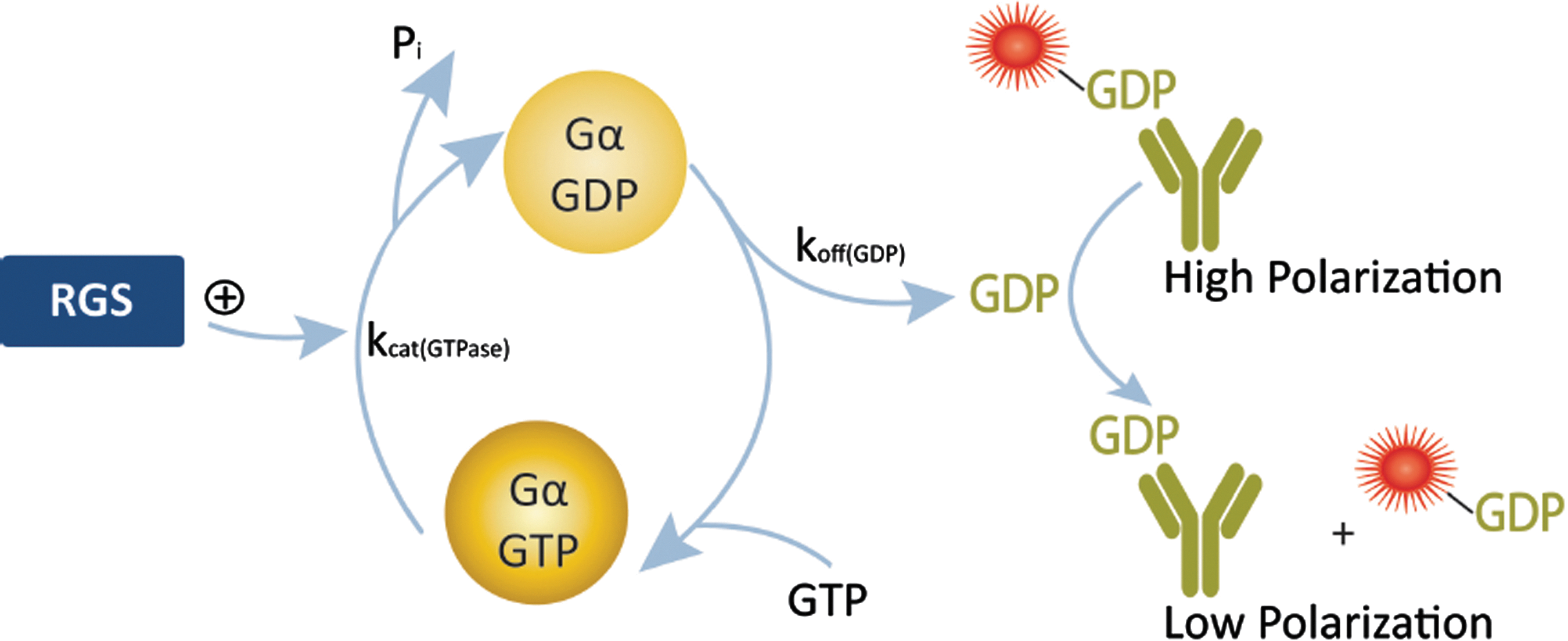
The Transcreener technology can be leveraged into additional target families using approaches such as coupling enzymes, or in this case, mutated Gα proteins with altered GDP dissociation kinetics that allow direct detection of the GTPase-accelerating activity (GAP) activity of regulator of G-protein signaling (RGS) proteins using the Transcreener GDP Assay (J Biomol Screen 2009;14:1195–1206).
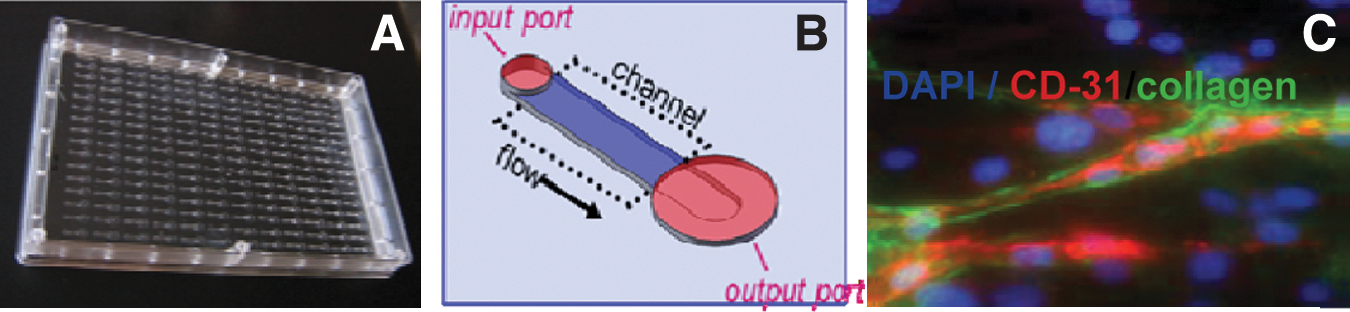
Can you please explain the iuvo™ Microconduit Array platform and how it is being used to enable high content assays?
The iuvo platform is an improvement upon multiwell plates, but instead of wells, iuvo plates have microchannels with different designs and patterns that enable users to do things they could not do in wells. The overall concept is to bring complex, highly miniaturized phenotypic assays into HTS. I thought about this a lot the first couple of years after starting BellBrook and read the work of some of the pioneers in the tumor microenvironment area such as Mina Bissell and Joan Brugge. I also talked a lot to a very smart biomedical engineer friend and colleague, Dave Beebe, about how to leverage microfluidics for this purpose.
There are obvious limitations to what you can do in a well, in terms of both biology and output. For instance, you cannot control cell positioning to allow you to look at paracrine signaling. You cannot tell the fibroblasts to stay in the right half of the well and the epithelial cells to stay in the left half. But positioning cells in a defined region using microfluidics and laminar flow is straightforward. People have known this for a long time and have experimented with building miniaturized tissue surrogates using microfluidics. The challenge was how to use such a microfluidic device in HTS because it requires an active pumping mechanism; we used to call it “the plumbing problem.” You can't have 384 tiny tubes running in and out of your plate.
The key enabling technology underlying iuvo, which came from a fortuitous discovery in Dave's lab, was the passive pumping technology, which uses the surface tension of water to drive fluid flow through the microchannels. You can displace the fluid in a microchannel simply by dispensing a drop of liquid, and with the right design the droplet will force itself through the channel. This discovery meant that arrayed microdevices could be integrated into an existing HTS instrumentation.
The concept behind iuvo is that instead of asking what you can do in a well, you ask what biology you want to replicate and what you want to learn about, and then you design a micro-conduit specifically for accomodating that biology and probing it as desired. Since high content analysis is the best platform for acquiring phenotypic assay data, we designed the plates to be compatible with high content platforms. An example of what you can do with iuvo is to run immunocytochemical protocols and do microscopic imaging of cells in a single plane in a three-dimensional extracellular matrix. The technology allows people to look at drug pharmacology and at cells in a three-dimensional matrix environment rather than in a cellular monolayer. That is an example of how the company is seeking to meet its goal of providing tools that enable people to incorporate more physiologically relevant biology into drug discovery.
I should add that BellBrook Labs recently signed an exclusive distribution agreement with Thermo Fisher for the first two iuvo products, the Chemotaxis Assay Plate and the Microchannel 5250. Thermo Fisher will be offering the iuvo plates to customers through its Cellomics ArrayScan platform.
Iuvo microconduit array technology.
Metabolic enzymes are finding a renewed focus as drug targets, not only in traditional areas involving metabolic disorders, but also as therapies for cancer and infectious diseases. Have you identified families of enzymes for which suitable assay formats are lacking but there exists interest in drug discovery?
The answer is yes and no. The metabolic enzymes being targeted for tumor hypoxia in cancer are the really boring ones people learned about in introductory biochemistry classes, enzymes such as hexokinase. These carbohydrate kinases are not a problem, but some of the more challenging targets are the oxidoreductases. There is still no method for detecting reduced versus oxidized nucleotides that is truly robust for HTS. There are ways to do it, but there are no really robust fluorescence or TR-FRET–based methods. Some of the lipid-metabolizing enzymes are also quite difficult, especially the microbial ones, because they tend to use obscure cofactors. We cannot invest a lot in assay development for a target that will only be screened by one or two companies.
One of the solutions we have come up with is to use coupling enzymes. An example would be acetyltransferases. We cannot directly detect coenzyme-A (coA), the product of acetyltransferases, but we can detect coA by coupling it to AMP formation, and we then detect AMP with a Transcreener assay.
The substrates for a metabolic or biosynthetic enzyme are often not readily available—for example, intermediates in purine metabolism. Does BellBrook Labs' technology offer solutions to this problem?
Yes, and the example of acetlytransferases relates to this. Another case would be methyltransferases. We had difficulty developing an antibody that specifically recognizes S-adenosylhomocysteine (SAH) versus S-adenosylmethionine, which differs by only a single methyl group. Our solution was to convert the SAH to AMP and detect the AMP.
For the biosynthetic enzymes, such as ligases, many of the targets—tRNA synthetases, for instance, which are involved in protein translation—are ATP-dependent ligases. They produce AMP and can be detected using the Transcreener AMP assay.
We have seen an evolution in profiling platforms, beginning as a collection of “key” targets (or anti-targets) against which pharmaceutical companies wished to assess the selectivity and specificity of their drug candidates, to more focused gene family arrays, particularly kinases. Do you think there will be comparable drivers and efforts to consolidate metabolic enzymes or other enzymes in a similar way? If so, what are the barriers to assembling these types of profiling platforms?
Definitely. About 15% of the coding capacity of the human genome is for proteins that bind purine nucleotides. There is no straightforward approach for predicting chemo-selectivity of different functional classes of enzymes based on their nucleotide-binding domains. But there is a very good chance that a compound that binds to a kinase ATP site with nanomolar affinity will interact with other ATP-utilizing enzymes, most of which are not currently included in profiling panels. There are all kinds of ATP-utilizing enzymes, such as ligases, helicases, and others that are not generally looked at. I think that using broader panels that include other enzymes besides kinases would yield a much more accurate picture of compound selectivity, give insights into mechanisms of action, and provide a new way to leverage the tremendous investment and knowledge in kinase chemoselectivity to other types of targets. I am all for broader selectivity profiling.
There really are no big technical barriers. The enzymes can be produced and the assay methods are available. The question is, do pharmaceutical companies want to broaden the scope of their profiling activities and look at how dirty their compounds really are? There are positive and negative aspects to multi-target activity by a single compound, and there may be benefits to understanding which currently available drugs hit multiple targets, which of those are involved in the drug's mechanism of action, and which are involved in its side effects, leading to a better understanding of how to take a more systems-based approach to drug discovery.
There has been a long-standing debate between molecular target-based reverse-chemical genomics approaches versus phenotypic forward-genetic approaches in drug discovery. Improved downstream target identification techniques are increasingly enabling phenotypic assays. How do you see BellBrook Labs' strategy evolving to address the needs in both isolated target-based assays and cell-based phenotypic assays?
It really depends on your goals. Are you trying to improve on an existing drug with a defined molecular mechanism of action? If so, then screening a known target often makes the most sense. You might be looking at modifications to that mechanism of action, for instance a longer residence time. But if you are trying to develop a first-in-class small molecule, then phenotypic screening casts the broadest net, so there are situations in which you might want to use that approach. In fact, retrospective studies have shown that the majority of first-in-class-drugs over the past several years were discovered using phenotypic screening methods.
As genomics research and related approaches reveal growing numbers of drug targets, what strategies can be used to prioritize them for assay development?
The best anecdotal example would be microbial targets. Genomics has revealed a tremendous number of microbial targets, and a number of pharmaceutical companies have made a huge investment in exploiting microbial genomic targets. They did it in a smart way, establishing large screening programs. But they came up with very little. In the end, their conclusion was that it is better to stick with the existing target classes and to work on the chemistry. A group from GlaxoSmithKline wrote a paper a few years ago (Nat Rev Drug Disc 2007;6:29–40) summarizing this work.
What does that tell you? Consider that the original antibiotics were discovered not by screening against molecular targets, but by screening for microbial viability, using a phenotypic screen. So the best way to prioritize genomic targets to identify a first-in-class compound is to do a phenotypic screen.
Is an increase in participation in collaborations part of your company's strategy for developing novel assays?
Absolutely. During the past year we have started an assay development and profiling service using the iuvo platform for applications such as tumor cell invasion and cell chemotaxis. But moving beyond this, one of the core capabilities we have established for the iuvo platform is the ability to do rapid design and prototyping. We are actively looking for opportunities to leverage the iuvo platform to develop additional assays based on customer needs, not only using existing iuvo plates, but also designing and fabricating customized plates to address specific questions that our partners want to answer. And we are making an effort to align this approach with our grant-funded research. For instance, we are seeking partners who are interested in air–liquid interface cultures, for things like bronchial epithelia or skin models, and this is an area in which we have an active grant-funded R&D program.
In recent years there has been a surge of assay development and HTS activity in academic and nonprofit labs. Have you needed to adopt a different approach with these labs as compared with those in pharmaceutical companies? Are there specific needs, capabilities, or goals of academic labs that guide product development targeting these users?
The academic screening labs function very much like pharmaceutical HTS laboratories, except they are generally looking at more diverse targets. The National Institutes of Health is not funding a lot of kinase screens; it wants to see more diverse targets and new targets. Transcreener is useful for that because it can be used for a large array of targets. As far as our overall approach, we do a lot of up-front collaboration with pharmaceutical and biotechnology partners, as well as with our academic partners, bringing their targets in-house and validating them in our assays.
From the perspective of the mission and goals of the BellBrook resource center, why do you believe it is so important to educate the user on the basis and components of the assay technology?
That relates to the philosophy of our company from the start. We have taken a very collaborative approach. We get to know our customers very well, many on a personal basis, and I think they view us as a resource. Despite the fact that HTS has now been a dominant drug discovery technology for nearly 20 years, getting an enzyme to work in an assay can still be a challenge. We are very good at it, and our customers appreciate that. We are able to take their enzymes and incorporate them into our assays very quickly. Part of that involves helping them directly, and part is educating them so they will be more successful in achieving their goals and in using our products.
In this regard, we are starting to produce some enzymes internally for some of the emerging target areas that are addressed by Transcreener assays, such as GTPases, because we have seen a fair number of customers have to terminate screening programs due to lack of high quality enzymes. This is just a shame, because it is preventing the exploration of new targets, but it is the reality of pharma drug discovery. We want to do whatever we can to remove the barriers to incorporating a broader swath of human biology into drug discovery.