Zhang M, Jiang XQ, Le HN, Wang P, Ye BC: Dip-and-read method for label-free renewable sensing enhanced using complex DNA structures. ACS Appl Mater Interfaces 2013;5:473–478.
Abstract: A label-free assay is reported in this work for the detection of DNA with enhanced sensitivity using complex DNA structures (DNA tetrahedrons) based on the biolayer interferometry. The DNA tetrahedrons help to amplify the optical signals of the biolayer interferometry, thus improving the detection limit of DNA by about 100-fold. We further demonstrated that this method could be expanded to ATP detection by taking advantage of the target-dependent adaptability of aptamers. It appears to us that this new label-free assay promises new opportunities for developing novel biolayer interferometry assays.
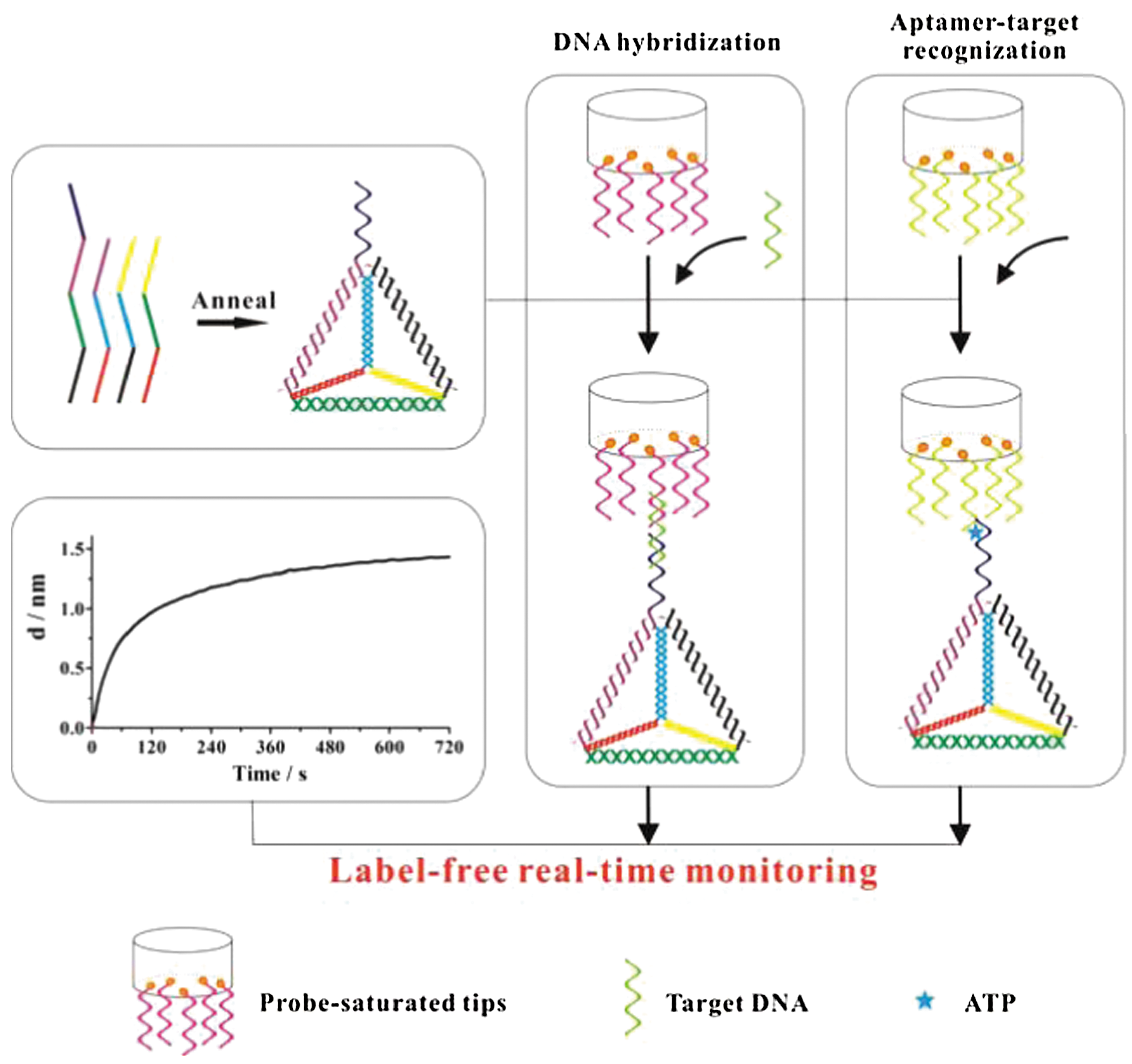
Commentary:Biolayer interferometry (BLI) is an optical technique that measures biomolecular interactions in real time based on interference patterns of white light reflected from immobilized ligand surfaces. Binding between an analyte in solution and a ligand immobilized on biosensor tips subsequently induces a wavelength shift in the interference pattern, which corresponds to the increase in optical thickness on the biosensor surface. This can be further processed to provide information on binding affinity and the kinetics of binding. The article by Zhang et al. presents a BLI assay with enhanced detection sensitivity though the incorporation of DNA tetrahedrons as probes in addition to those immobilized on fiber-optic tips. First reported by Goodman and coworkers, DNA tetrahedrons are designed to afford structural rigidity and stability through their triangulated architecture (Goodman et al., Science 2005;310:1661–1665). In this study, a DNA tetrahedron was synthesized through self-assembly of four DNA strands, and its successful assembly was validated by gel electrophoresis. It was further hypothesized that by capture of target molecules in solution through hybridization DNA tetrahedrons (see figure), the signal window and assay sensitivity could be substantially improved. In a subsequent proof-of-principle experiment, target DNA with concentrations in solution ranging from subnanomolar to 100 nM was measured with DNA probes immobilized on optical fiber tips in the absence and presence of DNA tetrahedron. Time-dependent signal response was easily observed at 1 nM target DNA when the DNA tetrahedron was present. Initial velocities of the hybridization response curves were further derived and were found to be linearly responsive with respect to the target DNA concentration. A 100-fold improvement in the limit of detection for the assay was estimated upon addition of the DNA tetrahedron. Using DNA oligos with various degrees of base mismatches, assay specificity was demonstrated through significant variations in the initial velocities captured from respective hybridization reactions. The assay was further extended for a successful ATP detection application using target-dependent aptamers as probe molecules. In both applications, regeneration procedures illustrated the reusability of the probe sensor. Using complex DNA structures such as DNA tetrahedrons, the method showed that sensitivity of BLI-based assays could be significantly improved for quantification of target molecules in solution. The method also holds potential as an alternative for genetic testing and point mutation screening purposes. Contributed by Wendy Lea.
Schematic illustration of the sandwich assay for label-free detection of sequence-specific DNA or ATP enhanced via a complex DNA structure (DNA tetrahedron), respectively.
Which Drug Combination will Unlock Triple-Negative Breast Cancer?
Ferraro DA, Gaborit N, Maron R, Cohen-Dvashi H, Porat Z, Pareja F, Lavia S, Lindzen M, Ben-Chetrit N, Sela M, Yarden Y: Inhibition of triple-negative breast cancer models by combinations of antibodies to EGFR. Proc Natl Acad Sci USA 2013;110:1815–1820.
Abstract: Breast tumors lacking expression of human epidermal growth factor receptor 2 (HER2) and the estrogen and the progesterone receptors (triple-negative; TNBC) are more aggressive than other disease subtypes, and no molecular targeted agents are currently available for their treatment. Because TNBC commonly displays EGF receptor (EGFR) expression, and combinations of monoclonal antibodies to EGFR effectively inhibit other tumor models, we addressed the relevance of this strategy to treatment of TNBC. Unlike a combination of the clinically approved monoclonal antibodies, cetuximab and panitumumab, which displaced each other and displayed no cooperative effects, several other combinations resulted in enhanced inhibition of TNBC's cell growth both in vitro and in animals. The ability of certain antibody mixtures to remove EGFR from the cell surface and to promote its intracellular degradation correlated with the inhibitory potential. However, unlike EGF-induced sorting of EGFR to lysosomal degradation, the antibody-induced pathway displayed independence from the intrinsic kinase activity and dimer formation ability of EGFR, and it largely avoided the recycling route. In conclusion, although TNBC clinical trials testing EGFR inhibitors reported lack of benefit, our results offer an alternative strategy that combines noncompetitive antibodies to achieve robust degradation of EGFR and tumor inhibition.
Commentary:Triple-negative breast cancer (TNBC) is an aggressive form of breast cancer that does not currently have any targeted therapeutics available. TNBC is characterized by the lack of expression of human epidermal growth factor receptor 2 (HER2 or EGFR2), estrogen receptor, and progesterone receptor, which are expressed in other types of breast cancers and can be targeted directly in those cases. TNBC cell growth is likely not fueled by estrogen, progesterone, or Her2 (the ligands for the three absent receptors), and therefore hormonal therapy or anti-Her2 treatments are likely to be ineffective. Current TNBC drug treatments consist primarily of standard systemic chemotherapies. TNBC patients respond differently to the various current treatments, and this indicates that the drivers of cancer for TNBC patients may differ between patients. TNBC cells from some patients appear to lack the ability to repair DNA damage efficiently, so platinum-containing drugs that cause DNA damage are being investigated in several clinical trials. About 50% of TNBC cells express epidermal growth factor receptor (EGFR), and indeed clinical trials have been undertaken to target EGFR with antibodies or tyrosine kinase inhibitors or with combinations of an anti-EGFR antibody and a standard chemotherapeutic agent. There are currently many ongoing trials, which predominantly focus on combinations of agents to treat TNBC. The authors here have investigated antibodies against EGFR that can be used concurrently and lead to synergy. They were able to find two EGFR antibodies (panitumumab and mAb111) that are noncompetitive for binding to EGFR (i.e., binding of one does not preclude binding of the other). The combination increased EGFR ubiquitination, which is often associated with receptor degradation (see first figure). Additionally, pretreatment with bortezomib or bafilomycin before treatment of the antibody pair was used to try to tease out mechanism. Degradation was inhibited by the inhibitor of lysosomal hydrolases (bafilomycin) but not by an inhibitor of the proteasome degradation system (bortezomib; see first figure, panel C). The effect of single agents versus combinations was also studied (see second figure). EGFR was degraded by combinations of antibodies but not by the single agents. Additionally, a 50% reduction in cell migration (via an invasion assay) was observed in combination but not with single agents. The authors point out that there is an ongoing clinical trial for two noncompetitive antibodies that target EGFR but for different indications (squamous cell carcinoma of the head and neck and metastatic colorectal cancer). It will be quite interesting to see whether this combination is effective for those cancer types and by extension may be worth pursuing for TNBC. TNBC is an important target representing ∼15%–20% of breast cancer cases and finding the right combination of drugs to increase overall survival is imperative. The authors have provided a good case for trying this new combination of EGFR antibodies in the TNBC patient population who have high levels of EGFR expression. Contributed by Mindy I. Davis.
Anti-EGFR antibodies enhance receptor ubiquitination and degradation. (A) Serum-starved HeLa cells were incubated with mAbs (10 μg/mL), a combination (each at 5 μg/mL), or with EGF (10 ng/mL), and lysates analyzed using immunoprecipitation (IP) and immunoblotting (IB). (B) HeLa cells transfected with a plasmid encoding an MYC peptide–tagged CBL, or an empty vector, were serum-starved and treated for 3 h with the indicated mAbs (20 μg/mL) or a combination (each at 10 μg/mL). Alternatively, cells were treated for 10 min with EGF (10 ng/mL). Lysates were probed as indicated. (C) HeLa cells that were preincubated (12 h) with bortezomib (2 μM) or bafilomycin (10 nM) were incubated (60 min) with EGF (10 ng/mL) or for 6 h with the indicated combination of mAbs (each at 10 μg/mL). Lysates were subjected to IB and signal quantification. EGF, epidermal growth factor; EGFR, EGF receptor; mAbs, monoclonal antibodies.
A combination of antibodies down-regulates EGFR and inhibits invasion of TNBC cells. (A) Whole extracts were prepared from the indicated cell lines after treatment with EGF (10 ng/mL, 1 h) or with the mAbs (20 μg/mL total, 6 h). Lysates were immunoblotted as indicated. (B) BT-549 cells were treated for 48 h with mAbs (20 μg/mL total) and lysates immunoblotted for EGFR and ERK2. (C, D) BT-549 cells were treated with mAbs as in (B). Thereafter, cells were plated in the upper compartment of invasion chambers. The lower compartments were filled with the respective mAb-containing media. Eighteen hours later, the filters were removed, fixed, permeabilized, and stained with methyl violet (0.3%). Cells growing on the upper side of the filter were removed and cells on the bottom side were photographed and quantified.
Nmr Mixes
Wu B, Zhang Z, Noberini R, Barile E, Giulianotti M, Pinilla C, Houghten RA, Pasquale EB, Pellecchia M: HTS by NMR of combinatorial libraries: a fragment-based approach to ligand discovery. Chem Biol 1993;20:19–33.
Abstract: Fragment-based ligand design (FBLD) approaches have become more widely used in drug discovery projects from both academia and industry, and are even often preferred to traditional high-throughput screening (HTS) of large collection of compounds (>105). A key advantage of FBLD approaches is that these often rely on robust biophysical methods such as NMR spectroscopy for detection of ligand binding, hence are less prone to artifacts that too often plague the results from HTS campaigns. In this article, we introduce a screening strategy that takes advantage of both the robustness of protein NMR spectroscopy as the detection method, and the basic principles of combinatorial chemistry to enable the screening of large libraries of fragments (>105 compounds) preassembled on a common backbone. We used the method to identify compounds that target protein-protein interactions.
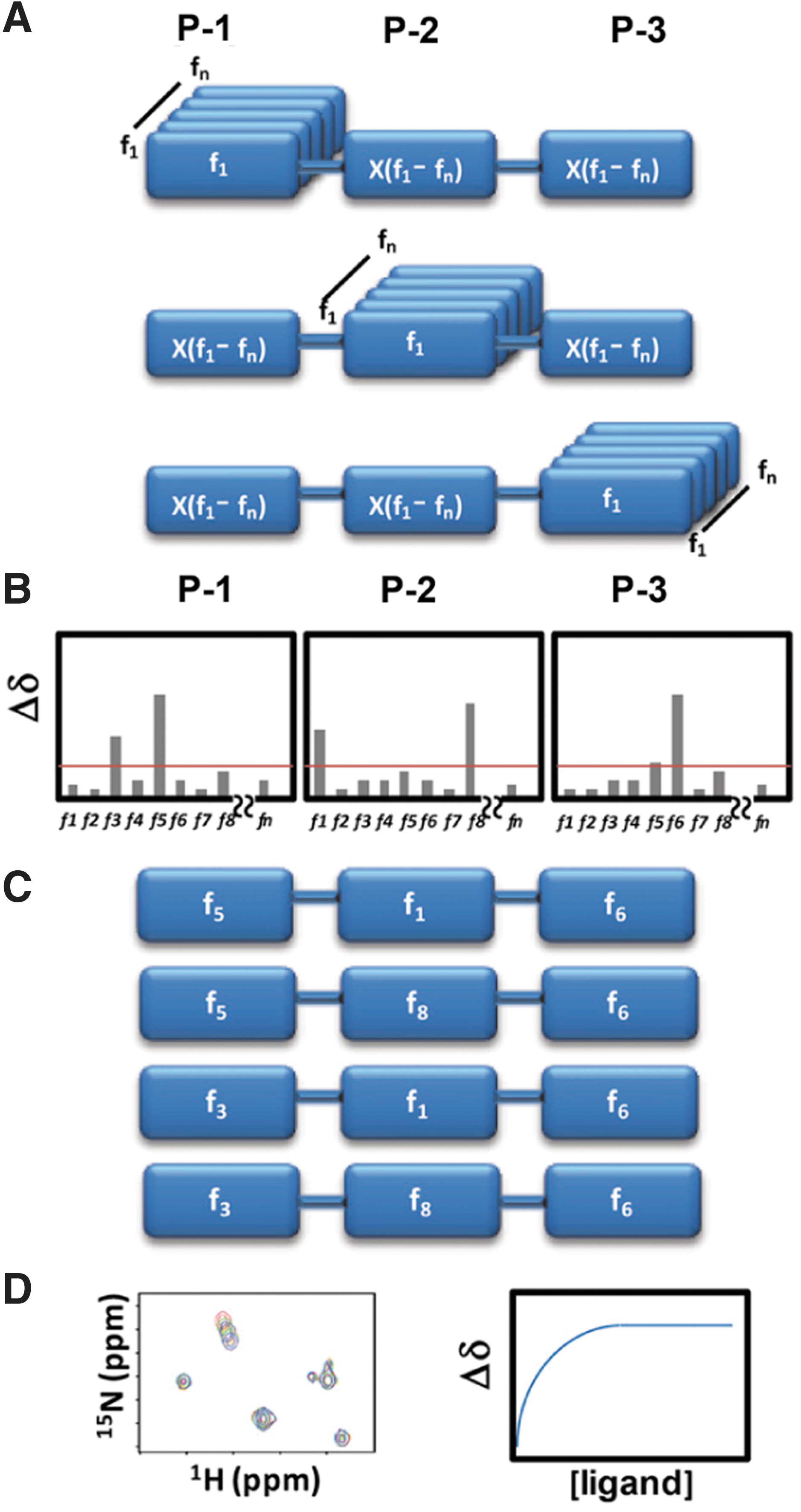
Commentary:Nuclear magnetic resonance (NMR) applied to drug discovery has largely been in the secondary assay stages of compound optimization, in which low numbers of compounds are characterized. Structure–activity relationships obtained using NMR (SAR by NMR) have been used to drive discovery efforts using small compound “fragments” (typically <200 MW) to identify weak binders to a protein that can be used as starting points for construction of higher affinity compounds. This article describes a way to use NMR to screen large (>105 compounds) collections of compounds in a high-throughput manner. To achieve this, the authors combine the rationale of combinatorial chemistry with the benefits of NMR measurements, wherein ligand–protein interactions can be directly measured. Large combinatorial libraries can easily achieve sizes of ∼105 compounds, but screening this many compounds by NMR would be unrealistic due to the time it takes to collect an NMR spectrum. One approach to reduce the number of samples to be tested from combinatorial collections involves the use of a positional scanning library. A positional scanning library reduces the number of samples that need to be tested by providing mixtures of compounds with one position fixed. For example, a combinatorial library that varies three positions with 100 synthons at each position gives a combinatorial library size of 100 × 100 × 100 = 106 samples that require testing. However, in the positional scanning approach, only 300 mixtures are screened, with each mixture fixing one fragment position and containing all possible combinations of fragments at the other two positions. Therefore, instead of testing 106 samples, only 300 samples need to be tested (“HTS by NMR”; see figure). In the NMR experiment, each individual compound in the mixture is present at very low concentrations (∼nM), which is below the limit of detection for NMR. However, the fixed fragment should have a concentration equal to the total concentration of the compound in the experiment. Using a 1 mM concentration of the mixture and assuming a 1% hit rate, one could expect a fragment that binds to the target to have an effective concentration of 10 µM, which is on the order of the protein concentration used in the NMR experiment (50 µM). To test this system, the BIR3 domain of the antiapoptotic protein XIAP, known to bind to the N-terminus of caspase-9 and prevent apoptosis, was used because there are known ligands that can disrupt this interaction, such as the high-affinity tetrapeptide ligand AVPF. Previous studies have shown that Ala and Pro in the tetrapeptide motif are absolutely required for binding. To test if this interaction could be found using HTS by NMR, a combinatorial library was screened in mixtures where either the P1 position was fixed with an Ala residue or the P3 position was fixed with a Pro residue. A series of 2D [15N, 1H] heteronuclear single quantum correlation (HSQC) experiments were performed using 15N-labeled BIR3 in the absence and presence of 1 mM mixtures. Using peptides of four residues or longer, the HSQC experiments supported the prior work, demonstrating a requirement of Ala and Pro at the P1 and P3 positions, respectively. HTS by NMR was then used to discovery new ligands to the ligand-binding domain of the EphA4 receptor tyrosine kinase. The HTS by NMR approach described here offers a method to rapidly screen large libraries of preassembled fragments using a method where one measures binding of a compound to a target. This combination of combinatorial chemistry and an analytical assay method should enable the discovery of leads for many target classes which are amenable to analysis by NMR. Contributed by Doug Auld.
Schematic representation of the HTS by NMR approach. (A) First, a positional scanning library of compounds needs to be assembled. In the example, a three-position synthetic combinatorial library is prepared. With a library of n fragments, there will be 3 × n mixtures, each containing n × n compounds. Hence, rather than synthesizing and testing n × n × n individual compounds, the approach would result in testing 3 × n mixtures. For example, a library of 100 fragments assembled at three different positions could be sampled by screening 300 mixtures (100 + 100 + 100), rather than by synthesizing and testing 1,000,000 (100 × 100 × 100) individual compounds. (B) Chemical shift perturbations induced by each mixture are measured and reported as function of the fixed fragment at each position. (C) Based on a determined Δδ cutoff, preferential fragments for each position are selected and final individual compounds are synthesized. (D) The binding affinity of the synthesized compounds is then determined via 2D heteronuclear NMR titration experiments.
Protein Stability Sandwich
Hailu TT, Foit L, Bardwell JCA: In vivo detection and quantification of chemicals that enhance protein stability. Anal Biochem 2013;434:181–186.
Abstract: We have devised protein-folding sensors that link protein stability to TEM-1 β-lactamase activity. The addition of osmolytes and other compounds with chemical chaperone activity to the growth medium of bacteria containing these sensors increases β-lactamase activity up to 207-fold in a dose-dependent manner. This enables the rapid detection and sensitive quantification of compounds that enhance in vivo protein stability.
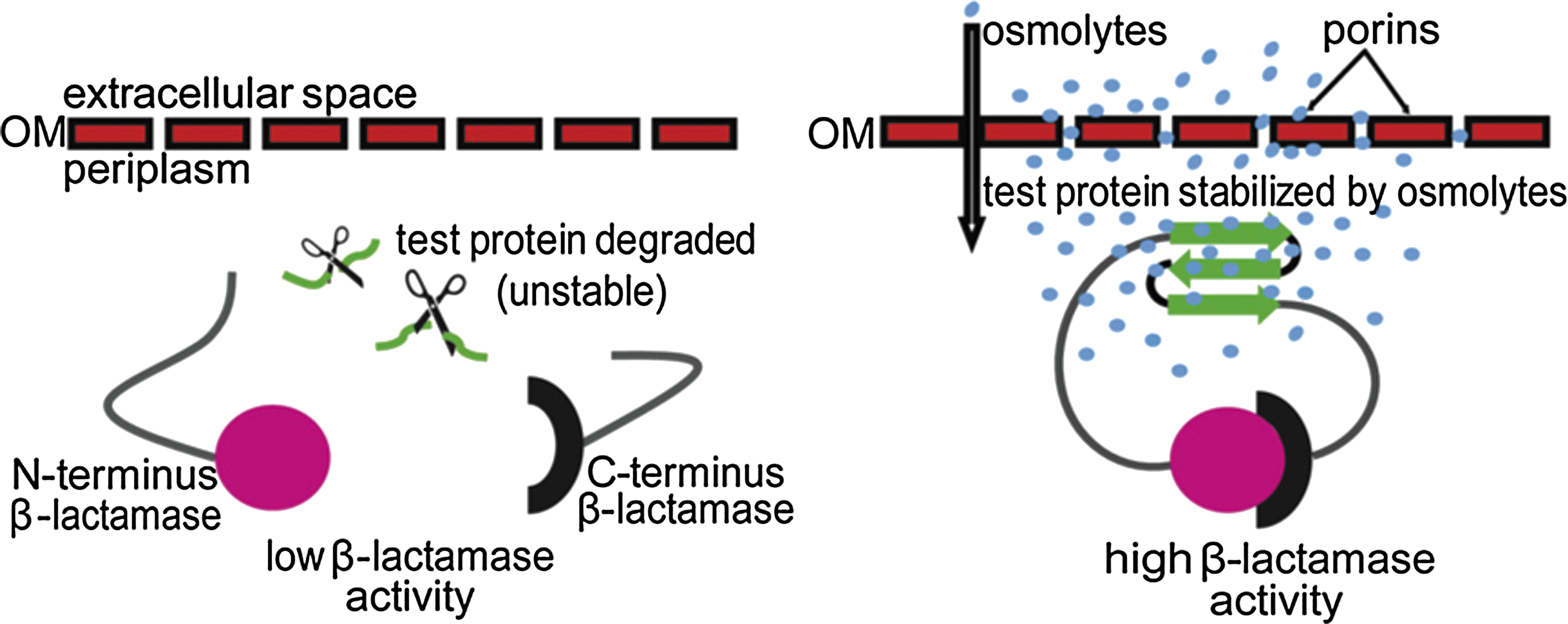
Commentary:Ligand binding to proteins is often coupled with thermodynamic stabilization of protein structure. This has been exploited using biophysical approaches such as differential scanning fluorescence, wherein increases in protein thermal melting points are monitored to confirm ligand binding to a protein. Thermodynamic stabilization of proteins in cells by a ligand can also slow the degradation of the target protein, leading to a rise in cellular protein levels. This article takes advantage of this general property to construct a cell-based sensor of protein stability. For this purpose, β-lactamase was used, and a test protein was inserted between residues 196 and 197 of the β-lactamase enzyme. This β-lactamase “sandwich fusion” protein exhibits a cell-based half-life that depends on the stability of the test protein. When the test protein is intact, the two β-lactamase fragments remain in close proximity, and an active enzyme is generated so that the amount of β-lactamase activity can be used as a measure of the test protein stability (see figure). To pilot this system, the authors used a known unstable protein mutant (the I22V mutant of immunity protein 7 [lm7]). The lm7 variant is destabilized by 8.9 kJ/mol relative to the wild-type (WT) protein. To test for stabilizers, the β-lactamase sandwich fusion protein was expressed in the periplasm of gram-negative bacteria. Due to the presence of porins, the periplasm is permeable to compounds <600 Da. In the assay, various osmolytes were tested, and several of these showed large increases in β-lactamase activity supporting stabilization of the fusion protein. The authors confirmed that the β-lactamase activity increases were due to increased protein levels by Western blotting using anti–β-lactamase antibodies. The stabilization appeared to be specific for the lm7 test protein because Western blots showed much larger stabilization with the fusion protein compared to WT β-lactamase alone. For example, sucrose showed a 16.5-fold increase in protein levels using the β-lactamase lm7 I22V fusion, but only a 1.9-fold increase using the bacterial strains expressing WT β-lactamase. Further, no increases in β-lactamase mRNA levels were found in the presence of osmolytes. Some surprises were found in the study. For example, urea was found to stabilize the fusion protein in bacterial cells at a concentration of 0.2 M. Urea, a common protein denaturant, is not expected to be able to stabilize protein structure, but some reports have suggested that protein denaturants act to enhance H-bonding and residue packing when used at sub-denaturant concentrations. The system described here can be used to identify ligand binding to a target protein in a cell-based format, provided that the fusion protein allows for reconstruction of the split β-lactamase. Such an assay may identify compounds with unique mechanisms of action. Contributed by Doug Auld.
Schematic representation of a sandwich fusion system for assessing the influence of osmolytes on protein stability in vivo. An unstable test protein (green) is inserted into β-lactamase in the form of a sandwich fusion. In the absence of osmolytes, the inserted protein is subject to degradation by periplasmic proteases, symbolized by scissors. This results in a separation of the two β-lactamase fragments and, hence, decreased β-lactamase activity. Osmolytes (blue circles) diffuse into the periplasm through the holes generated in the outer membrane (OM) by endogenous porins. The presence of osmolytes favors a stable fold of the inserted protein. This allows an association of the N fragments (magenta circle) and C fragments (black semicircle) of β-lactamase, resulting in high enzymatic activity.
Bacteria Reprogram Host Cells in Order to Survive and Spread
Masaki T, Qu J, Cholewa-Waclaw J, Burr K, Raaum R, Rambukkana A: Reprogramming adult Schwann cells to stem cell-like cells by leprosy bacilli promotes dissemination of infection. Cell 2013;152:51–67.
Abstract: Differentiated cells possess a remarkable genomic plasticity that can be manipulated to reverse or change developmental commitments. Here, we show that the leprosy bacterium hijacks this property to reprogram adult Schwann cells, its preferred host niche, to a stage of progenitor/stem-like cells (pSLC) of mesenchymal trait by downregulating Schwann cell lineage/differentiation-associated genes and upregulating genes mostly of mesoderm development. Reprogramming accompanies epigenetic changes and renders infected cells highly plastic, migratory, and immunomodulatory. We provide evidence that acquisition of these properties by pSLC promotes bacterial spread by two distinct mechanisms: direct differentiation to mesenchymal tissues, including skeletal and smooth muscles, and formation of granuloma-like structures and subsequent release of bacteria-laden macrophages. These findings support a model of host cell reprogramming in which a bacterial pathogen uses the plasticity of its cellular niche for promoting dissemination of infection and provide an unexpected link between cellular reprogramming and host–pathogen interaction.
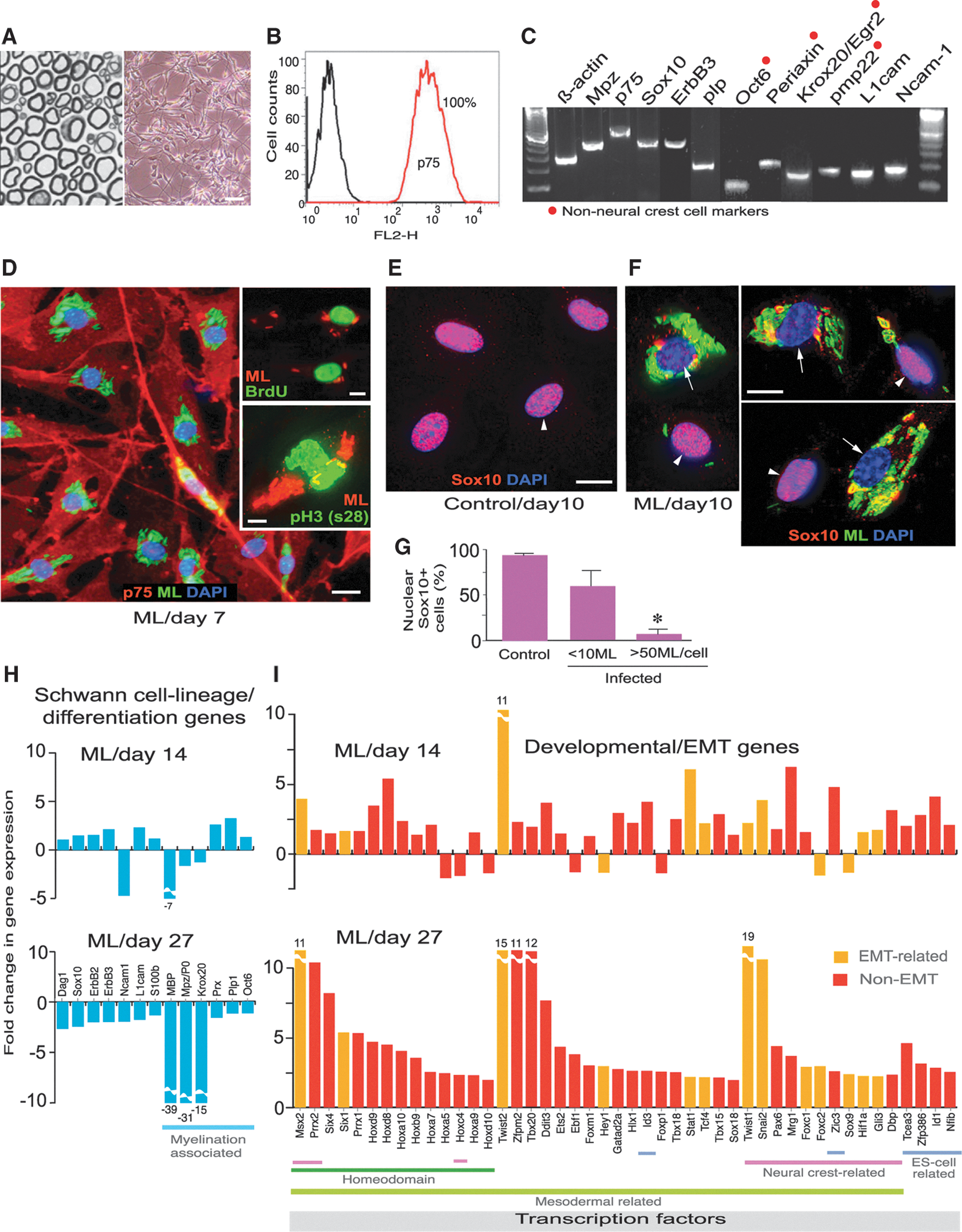
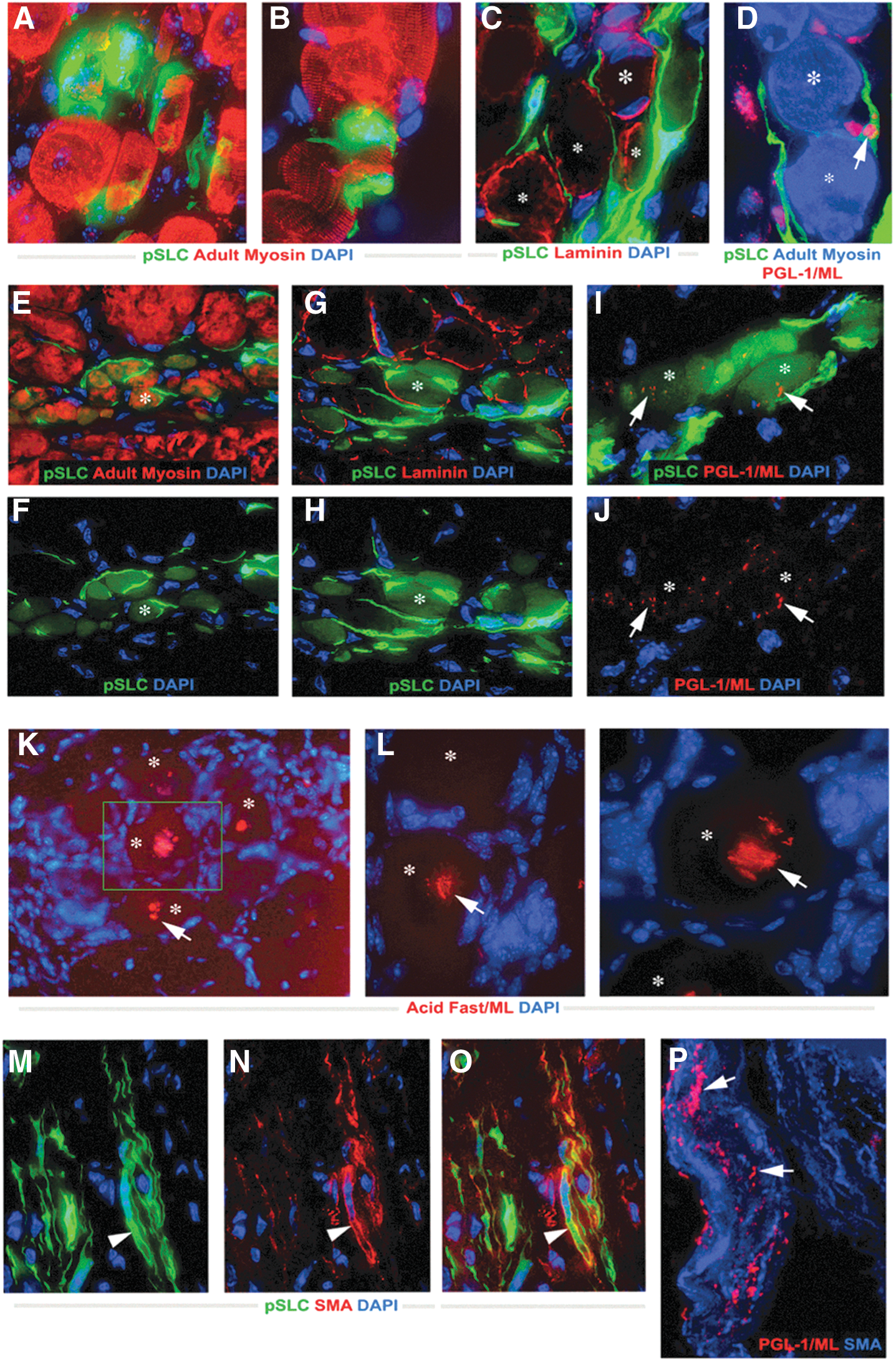
Commentary:The bacterium causing leprosy, Mycobacterium leprae, has been next to impossible to study because of extreme difficulties associated with its culturing and the lack of animal models. A particularly vexing question that remains unanswered is how M. leprae, an obligate intracellular pathogen, inflicts damage to separate tissues and organs around the body. The present study provides a twofold breakthrough in this field. The authors were not only able to create a model for the infection using mouse Schwann nerve cells, but in the course of their studies the team also discovered a fairly unexpected mechanism by which the mysterious bacteria invade human tissues. Through incubation of isolated Schwann cells with M. leprae, the authors found that the bacteria trigger reprogramming of the adult nerve cells, turning them to a progenitor/stem-like type (first figure). These cells, in turn, can detach from their typical structures comprising the sheaths of motor and sensory neurons and, given their plasticity and newly acquired mesoderm characteristics, migrate to distal sites and re-establish a site of infection and tissue damage through redifferentiation. This mechanism was verified by the authors through in vitro testing using co-culturing and in vivo testing using a nude mouse model (second figure). Although it still remains to be established how exactly the leprosy bacteria trigger the Schwann cell reprogramming, the new paradigm of bacterial colonization and spreading may have wide implications for our understanding of cell differentiation, tissue plasticity, and host–parasite interactions. Contributed by Anton Simeonov.
Adult peripheral nerve-derived Schwann cells undergo reprogramming events in response to intracellular Mycobacterium leprae (ML). (A, B) Methylin blue–stained semithin section of adult nerves (A,left) from which Schwann cells were isolated (phase image) (A,right) and purified by FACS with anti-p75 antibody (B). (C) RT-PCR of purified p75+ Schwann cells. Highlighted genes (red dots) are late Schwann cell developmental and myelin markers that are not present in neural crest cells. (D) Purified Schwann cells were infected with ML, fixed at day 7, and immunolabeled with anti-p75 (red) and anti–PGL-1 (iML in green) antibodies, counterstained with DAPI (blue). Insets: BrdU uptake (green, top) and antibody to phospho-histone H3 (Ser28; green, bottom) double-labeled with PGL-1 antibody (red). (E–G) Expression of nuclear Sox10 in control (E) and infected (F) Schwann cells at day 10 post-infection; antibodies to Sox10 (red) and PGL-1 (iML; green), counterstained with DAPI (blue) (F). (F) Selected representative Deltavision images from infected cultures with high (arrows) and low bacterial loads (arrowheads). (G) Quantification of nuclear Sox10 in control and infected Schwann cells with iML per cell. (H, I) Gene array analyses at days 14 and 27 post-infection. Schwann cell lineage/differentiation-associated genes (H) and development- and EMT-associated transcription factor (TF) genes (I) are shown as fold change in expression. See also the article's Supplemental Information. Scale bars: (D) 50 μm, (F, G) 10 μm. RT-PCR, reverse transcription polymerase chain reaction; DAPI, 4′,6-diamidino-2-phenylindole,dihydrochloride.
Redifferentiation of reprogrammed progenitor/stem-like cells (pSLC) contributes to passive bacterial transfer to skeletal muscles and smooth muscles in vivo. (A–D) Transverse frozen sections of tibialis anterior (TA) muscles injected with ML-infected GFP+ pSLC after 10 days. Deltavision images showed GFP + pSLC (green) either fused with myofibers (A, B) or migrating in between muscle fibers (C, D). Antibodies to adult myosin detected muscle fibers (red in A,B;blue in D) and laminin demarcated individual muscle fibers (red in C). Antibodies to PGL-1 detected ML (in red; arrow) associated closely with muscle fibers (D). Asterisks in all figures denote individual myofibers. (E–J; Deltavision images) Infected GFP+ pSLC were incorporated into regenerating muscles at 3 weeks post-injection. GFP + myofibers are positive for adult myosin (E, F) and are demarcated by laminin (G, H). pSLC incorporation passively transmitted ML to myofibers as detected by PGL-1+ ML (red; arrows) in GFP+ myofibers (I, J). (K, L) Bacterial presence within the skeletal muscle fibers (asterisks) was confirmed by acid fast mycobacterial staining (red). Arrows show clumps of rod-shaped ML in several individual muscle fibers. (M–P) Administered infected GFP+ pSLC differentiate into SMA+ smooth muscles in the dermal area. Antibody to SMA (red) colocalizes with differentiated GFP+ pSLC (green; arrowheads); merged image is shown in (O). (P) Higher magnification showed the PGL+ ML (red; arrows) within SMA+ smooth muscles (blue) near dermal vessels. Magnifications: (A–D, K, L) 60 × ; (E–J, M–O) 40 × ; (P) 100×.
A Step Closer to Personalized Medicine
Kamiyama H, Rauenzahn S, Shim JS, Karikari CA, Feldmann G, Hua L, Kamiyama M, Schuler FW, Lin M, Beaty RM, Karanam B, Liang H, Mullendore ME, Mo G, Hidalgo M, Jaffee E, Hruban RH, Jinnah HA, Roden RBS, Jimeno A, Liu JO, Maitra A, Eshleman JR: Personalized chemotherapy profiling using cancer cell lines from selectable mice. Clinical Cancer Res 2013;19:1139–1146.
Abstract:
Purpose. High-throughput chemosensitivity testing of low-passage cancer cell lines can be used to prioritize agents for personalized chemotherapy. However, generating cell lines from primary cancers is difficult, because contaminating stromal cells overgrow the malignant cells.
Experimental Design. We produced a series of hypoxanthine phosphoribosyl transferase (hprt)–null immunodeficient mice. During growth of human cancers in these mice, hprt-null murine stromal cells replace their human counterparts.
Results. Pancreatic and ovarian cancers explanted from these mice were grown in selection media to produce pure human cancer cell lines. We screened one cell line with a 3,131-drug panel and identified 77 FDA-approved drugs with activity, including two novel drugs to which the cell line was uniquely sensitive. Xenografts of this carcinoma were selectively responsive to both drugs.
Conclusion. Chemotherapy can be personalized using patient-specific cell lines derived in biochemically selectable mice.
Commentary:For many cancer patients, it is not clear at the outset which treatment will have the highest chance for success. Many cancer treatments have significant side effects, which can occur whether the drug provides any benefit to the patient or not. If it is possible to know at the outset the likelihood of treatment success for the various available drugs, the patient and doctor can choose a treatment course with the highest chance for success while minimizing the side effects and costs of ineffective treatments. Sometimes patients can be stratified on the basis of a marker from a biopsy of their cancer, but often they cannot, and it is not clear which treatments should be undertaken. Therefore, the process of finding the right drug is often trial and error. Ideally, a patient's cells could be tested ex vivo against the possible drugs to see which is most effective. When the patient's cells are initially removed and tested, the cells may be sick or dying and results of drug screening at this point could be misleading. The authors describe a method of expanding and stabilizing the human patient's cancer cells in a modified mouse (hypoxanthine phosphoribosyl transferase [hprt]–null immunodeficient mouse) and then removing those cells and testing the cells for drug sensitivity in an in vitro cell assay (see first figure). One of the important aspects of their research was that their modified mouse allows the noncancer fibroblasts of the human patient to be replaced by biochemically defective mouse cells, which can be selectively eliminated prior to the cell assay (by means of hypoxanthine, aminopterin, and thymidine [HAT]–containing media). In culture, the noncancer fibroblasts can overtake the cancer cells, so this method of stromal cell removal allows for the malignant cancer cells to be retained. This method was successful in seven of the nine cell lines attempted (six pancreatic ductal adenocarcinoma cell lines and one ovarian cancer cell line). One of the pancreatic lines (Panc502) was isolated from the mouse and used to test the in vitro sensitivity to a drug panel containing over 3,000 drugs (Johns Hopkins drug library panel, see second figure). The authors identified 10 drugs for further study that had not been shown to be active in pancreatic cancer cells previously. Nogalamycin and digitoxin were selective for Panc502 over untransformed or another transformed pancreatic duct line. Xenograft mice were then used to test the effect of the drugs, and there was a correlation between the in vitro response and the in vivo response. The authors point out that the current shortfall with this method was the time it took from initiation to completion of this study (8 months). They indicate that this timeline could be greatly reduced in the future by implanting additional mice, additional optimization of the procedure, and more automation of the drug screening process. It remains to be seen whether this timeline can be reduced to the point where it will be clinically useful for deciding treatment courses in a timely fashion. It can, though, be potentially useful even with the current timeline for generating cell lines for cancer types that currently do not have available lines. We know that there can be patient-to-patient variability on the effectiveness of drug treatments, so having more lines available for a given cancer type should be helpful for scientists trying to better understand the different disease drivers. Contributed by Mindy I. Davis.
Rapid pancreatic cancer cell line production. Samples explanted from Panc410 tumors grown in a nude hprt-null mouse in the presence (A–C) or absence (D–F) of HAT media, and photographed using phase microscopy at T = 0, 10 and 19 days in culture. Note that after 19 days in the absence of HAT media (F), fibroblasts have overgrown the culture. hprt, hypoxanthine phosphoribosyl transferase.
Chemosensitivity of a low-passage familial pancreatic cancer from surgery. Histogram of the number of drugs (frequency) as a function of % growth inhibition (A). Curve-fitting of Gaussian distribution onto the histogram (black line) distinguishes the distribution of drugs with little or no activity from those which demonstrate some level of activity above this distribution. Arrowhead indicates three standard deviations (+3 SD) above the mean. Arrows indicate the % growth inhibition for nogalamycin (N) and digitoxin (D). Cell line–specific sensitivity of nogalamycin and digitoxin in the cell lines Panc502, Panc486, and Panc410 (B). Values shown are the mean IC50 values of three replicates and error bars are the 95% confidence intervals. In vivo growth curves of subcutaneous mouse xenografted tumors raised from the Panc410 and Panc502 cell lines after treatment with nogalamycin, digitoxin, or control (C). ○, control; □, nogalamycin 0.2 mg/kg; ■, nogalamycin 1.0 mg/kg; △, digitoxin 0.4 mg/kg; ▲, digitoxin 2.0 mg. Normalized weight of tumors explanted from mice after 30 days of treatment (D). Normalized tumor weight of Panc410 and Panc502 in white columns or gray columns, respectively. Error bars are standard deviations. Fold changes between Panc410 and Panc502 are noted.
Growing Human Tumors in a Dish
Godugu C, Patel AR, Desai U, Andey T, Sams A, Singh M: AlgiMatrix™ based 3D cell culture system as an in-vitro tumor model for anticancer studies. PLoS ONE 2013;8:e53708.
Abstract:
Background. Three-dimensional (3D) in vitro cultures are recognized for recapitulating the physiological microenvironment and exhibiting high concordance with in vivo conditions. Taking the advantages of 3D culture, we have developed the in vitro tumor model for anticancer drug screening.
Methods. Cancer cells grown in 6- and 96-well AlgiMatrix™ scaffolds resulted in the formation of multicellular spheroids in the size range of 100–300 µm. Spheroids were grown in two weeks in cultures without compromising the growth characteristics. Different marketed anticancer drugs were screened by incubating them for 24 h at 7, 9, and 11 days in 3D cultures and cytotoxicity was measured by AlamarBlue® assay. Effectiveness of anticancer drug treatments was measured based on spheroid number and size distribution. Evaluation of apoptotic and antiapoptotic markers was done by immunohistochemistry and RT-PCR. The 3D results were compared with the conventional 2D monolayer cultures. Cellular uptake studies for drug (doxorubicin) and nanoparticle (NLC) were done using spheroids.
Results. IC50 values for anticancer drugs were significantly higher in AlgiMatrix™ systems compared to 2D culture models. The cleaved caspase-3 expression was significantly decreased (2.09 and 2.47 folds, respectively, for 5-fluorouracil and camptothecin) in H460 spheroid cultures compared to 2D culture system. The cytotoxicity, spheroid size distribution, immunohistochemistry, RT-PCR, and nanoparticle penetration data suggested that in vitro tumor models show higher resistance to anticancer drugs and supported the fact that 3D culture is a better model for the cytotoxic evaluation of anticancer drugs in vitro.
Conclusion. The results from our studies are useful to develop a high throughput in vitro tumor model to study the effect of various anticancer agents and various molecular pathways affected by the anticancer drugs and formulations.
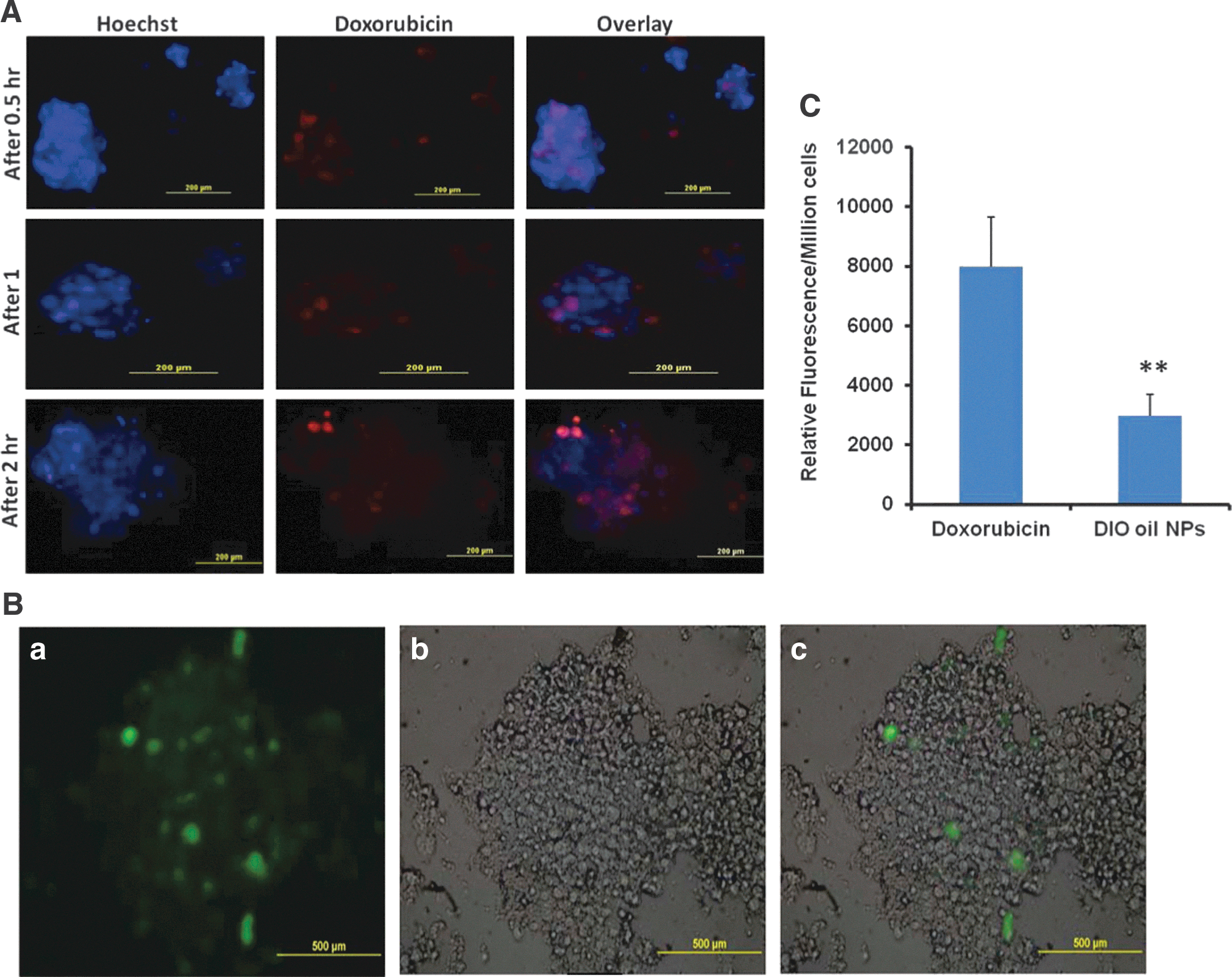
Commentary:Scientists are continually trying to improve the correlation between in vitro and in vivo drug efficacy. One avenue that has been explored is trying to modify cell culture techniques such that the in vitro cell assay environment more closely resembles the environment the cells encounter in the patient. One step towards that goal is moving from two-dimensional (2D) to three-dimensional (3D) in vitro cell culture because tumors in the body are not a single layer of cells attached to a microtiter plate, but rather are 3D shapes with distinct cell–cell and cell–matrix interactions. The authors here describe a culture technique in which tumor cells are grown in an alginate matrix to a size of 100–300 µm in 2 weeks. This time frame minimized the formation of necrotic lesions in the spheroids. The cell spheroids can be isolated by applying a nonenzymatic solution to dissolve the alginate scaffold. The technique was applied in both 6-well and 96-well assay plate formats. It will be interesting to see whether this can be scaled down further to 384-well format to increase the possibility of using this for large-scale screening campaigns. The authors measured IC50s for a variety of drugs in 2D and 3D culture systems of a lung tumor cell line and found that they were significantly higher in the 3D culture system (see table). The 3D culture is more resistant to anticancer drugs with a 10- to 60-fold shift in potency. This type of shift has been seen in other 3D systems as well, and current hypotheses for the shift include decreased penetration of the drug due to the formation of tight junctions, increased expression of E-cadherin, up-regulation of drug resistance genes or up-regulation of survival signaling pathways. Cleaved caspase-3 expression was markedly reduced in 3D culture, indicative of reduced apoptotic signaling. The H1650 cells that were used can be divided into parental and cancer stem cell (CSC) populations. The CSC populations grown in 2D normally require a special plate coating, but they readily grew in 3D. Indeed, the number of spheroid colonies from the CSC cells was higher than from the parental H1650 cells. Also, CSCs grown in 2D may lose some of their “stemness” properties. The authors went on to explore whether the differing IC50s could be due to differences in drug uptake. There was indeed a limited uptake of doxorubicin and nanoparticles by the spheroids (see figure), with just 10.52% and 3.41% found inside the spheroids, respectively. The nanoparticles were only able to penetrate the periphery. It will be interesting to have a large study comparing the different currently available 3D culture methods for a variety of cell types to see whether there may be a preferred method or whether the optimal method will be cell-line dependent. It will also be interesting to see whether these 3D methods can be reliably used in 384-well plates to further increase the likelihood of their adoption for high-throughput drug screening campaigns. While a spheroid of cells is still a long way from representing the complexity of a human being, even a small step towards decreasing the attrition of drugs through the discovery pipeline would be highly beneficial. Contributed by Mindy I. Davis.
Drug and nanoparticles uptake by spheroids. (A) Representative images of doxorubicin uptake in H1650 parental cell spheroids grown in 3D alginate scaffold system. Uptake images were taken at 0.5-, 1-, and 2-h time points. (B) Representative images of the nanoparticle uptake by H1650 parental cell spheroids. DIO oil was encapsulated in NLC and incubated with the spheroids for 2 h: (a) fluorescent image, (b) DIC image, and (c) merged image. The fluorescent image clearly indicates the nanoparticles uptake into spheroids. (C) Relative fluorescence intensities of free doxorubicin uptake and DIO oil–loaded NLC nanoparticles into 3D spheroids. Each data point is represented as mean ± SEM (n = 3). **p < 0.01 vs. doxorubicin group.
Comparative Analysis of IC50 Values of Various Anticancer Drugs in 2D and 3D Systems
Each data point is represented as mean ± SEM (n = 4–5).
aP < 0.001 vs. respective 2D groups.
G-Quadruplex Directly Tracked
Biffi G, Tannahill D, McCafferty J, Balasubramanian S: Quantitative visualization of DNA G-quadruplex structures in human cells. Nat Chem 2013;5:182–186.
Abstract: Four-stranded G-quadruplex nucleic acid structures are of great interest, as their high thermodynamic stability under near-physiological conditions suggests that they could form in cells. Here we report the generation and application of an engineered, structure-specific antibody employed to quantitatively visualize DNA G-quadruplex structures in human cells. We show explicitly that G-quadruplex formation in DNA is modulated during cell-cycle progression and that endogenous G-quadruplex DNA structures can be stabilized by a small-molecule ligand. Together these findings provide substantive evidence for the formation of G-quadruplex structures in the genome of mammalian cells and corroborate the application of stabilizing ligands in a cellular context to target G-quadruplexes and intervene with their function.
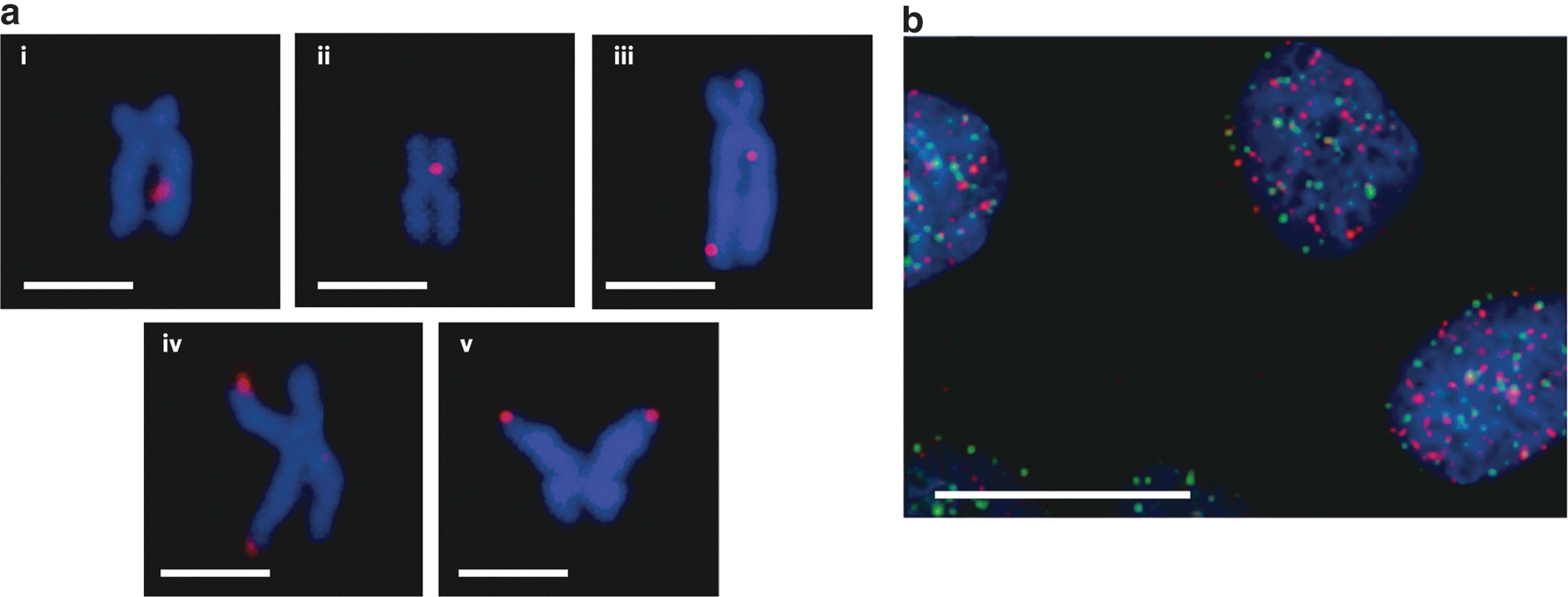
Commentary:G-quadruplexes have captured the fascination of many research groups during the past two decades. These unusual structures typically consist of four guanines conjoined through Hoogsteen hydrogen bonding into a planar configuration. The high stability of G-quadruplexes, attributed in part to the monovalent cation coordinated to the O6 lone pairs of each guanine, has led to various hypotheses regarding the role of these structures in regulation of transcription, telomere maintenance, and other cellular processes related to genomic DNA status. While biophysical studies of G-quadruplexes abound, tracking these structures inside the cell has been nearly impossible. Here, for the first time, a high-affinity detection agent is being provided that allows the visualization of G-quadruplexes in genomic DNA. Biffi and colleagues used phage display of single-chain Fv (scFv) fragments to identify the high-affinity monoclonal antibody BG4 which recognized G-quadruplexes at single-digit nanomolar affinity but at the same time did not bind to other DNA structures, as determined by competition enzyme-linked immunosorbent assay (ELISA). The new detection reagent was then used in a series of visualization experiments. Importantly, the authors showed that the antibody could be used to stain chromosomes and the initial studies revealed that G-quadruplexes were abundant at the ends of each chromosome, as previously postulated, and that in addition G-quadruplexes (seen as BG4 foci) were found interspersed throughout the genome (see figure). Moreover, use of the new antibody revealed that the level and distribution of G-quadruplexes change as the cell progresses through the stages of mitosis. The availability of this key detection reagent, in combination with modern tools such as the next-generation sequencing, should permit a precise mapping of the discrete sites of G-quadruplex structures, a step towards the eventual elucidation of their biological roles. Contributed by Anton Simeonov.
Localization of G-quadruplex structures in chromosomes. (a) Immunofluorescence for BG4 on metaphase chromosomes isolated from Hela cervical cancer cells. Discrete BG4 foci (red) were observed both within the nontelomeric regions (i–iii) and at the telomeres (iv, v), a well-characterized site of G-quadruplex formation. The symmetrical appearance of the foci in some sister chromatids (v) supports G-quadruplex formation within the same genomic locations in newly replicated DNA. Chromosomes are counterstained with DAPI (blue). Scale bars, 2.5 μm. (b) Absence of large co-localization between telomeric TRF2 proteins (green) and G-quadruplexes (red) in U2OS cells. This suggests that endogenous G-quadruplex structures are present largely outside the telomeres. Nuclei are counterstained with DAPI (blue). Scale bar, 20 μm. DAPI, 4′,6-diamidino-2-phenylindole.