
Abstract
Fibroblast growth factor 23 (FGF23) is a bone-derived endocrine key regulator of phosphate homeostasis. It inhibits renal tubular phosphate reabsorption by activating receptor complexes composed of FGF receptor 1c (FGFR1c) and the co-receptor Klotho. As a major signaling pathway mitogen-activated protein kinase (MAPK) pathway is employed. In this study, we established an FGF23-inducible cell model by stably expressing human Klotho in HEK293 cells (HEK293-KL cells) containing endogenous FGF receptors. To identify novel small molecule compounds that modulate FGF23/FGFR1c/Klotho signaling, we developed and optimized a cell-based assay that is suited for high-throughput screening. The assay monitors the phosphorylation of endogenous extracellular signal-regulated kinase 1 and 2 in cellular lysates of HEK293-KL cells after induction with FGF23. This cell-based assay was highly robust (Z′ factor >0.5) and the induction of the system is strictly dependent on the presence of FGF23. The inhibitor response curves generated using two known MAPK pathway inhibitors correlate well with data obtained by another assay format. This assay was further used to identify small molecule modulators of the FGF23 signaling cascade by screening the 1,280 food and drug administration-approved small molecule library of Prestwick Chemical. The primary hit rate was 2% and false positives were efficiently identified by retesting the hits in primary and secondary validation screening assays and in western blot analysis. Intriguingly, by using a basic FGF (bFGF)/FGFR counterscreening approach, one validated hit compound retained specificity toward FGF23 signaling, while bFGF signaling was not affected. Since increased plasma concentrations of FGF23 are the main cause of many hypophosphatemic disorders, a modulation of its effect could be a potential novel strategy for therapeutic intervention. Moreover, this strategy may be valuable for other disorders affecting phosphate homeostasis.
Introduction
Fibroblast growth factor 23 (FGF23) is a member of the fibroblast growth factor (FGF) family comprising 22 FGFs in humans. 1 Together with FGF19 and FGF21, FGF23 belongs to the FGF19 subfamily of endocrine FGFs, which shows a poor affinity for heparan sulfate (HS), and therefore can freely diffuse in the HS-rich extracellular matrix to enter into the bloodstream. 2 FGF23 is mainly released by cells derived from the osteoblast lineage. 3 While less is known about the production of FGF23 and its regulatory mechanisms in bone, most data focused on the endocrine function of FGF23 on target cells within the kidney. As a key regulator of phosphate homeostasis it reduces renal tubular phosphate reabsorption by lowering the expression of the sodium/phosphate co-transporters NaPi-2a and NaPi-2c in the proximal tubule of the kidney. 4,5 It has been demonstrated that excess actions of FGF23 result in several hereditary hypophosphatemic disorders including X-linked hypophosphatemia (XLH), 6 autosomal dominant hypophosphatemic rickets, 7 autosomal recessive hypophosphatemic rickets 1 and 2, 8,9 and in acquired conditions like tumor-induced osteomalacia. 10
Current medical treatment strategies include replacement therapies with oral administration of inorganic phosphate salts. 11 They have efficacy in managing and treating clinical symptoms, but renal phosphate wasting persists and the risk of side effects is high. 12 –15 Thus, there is an immense need for novel therapies with greater effectiveness and fewer side effects. One approach that directly targets FGF23 as a key effector of phosphate wasting has been described by Yamazaki et al. They generated monoclonal anti-FGF23 antibodies to neutralize excess actions of FGF23 in Hyp mice (murine homolog of XLH). 16 Due to the promising data in the animal model, a humanized antibody, KRN23, was developed, which ameliorated hypophosphatemia in patients with XLH after a single-dose and after monthly treatment. 17,18 Studies with known FGF receptor inhibitors, PD173074 and NVP-BGJ398, 19,20 and an inhibitor of mitogen-activated protein kinase kinase (MEK), PD0325901, 21 show diverse amelioration of the phenotype in Hyp mice. Another way to inhibit FGF23 actions is to suppress the binding of FGF23 to its FGF receptor/Klotho complex by competing with an isolated C-terminal fragment of FGF23. Injection of this C-terminal fragment increased serum phosphate levels both in wild-type and Hyp mice. 22 However, in contrast to the clinical trial with humanized anti-FGF23 antibodies none of the inhibitors or C-terminal fragment of FGF23 is yet under clinical investigations (for review see 23 ).
In this work, we propose a method to identify small molecules that regulate FGF23 signaling in a high-throughput screening (HTS) approach. The discovery of selective small molecule inhibitors of FGF23 signaling may provide the basis to manipulate this signaling pathway and would potentially validate it as a drug target in hypophosphatemic disorders.
Materials and Methods
Cell Culture
Adherent HEK293 cells were maintained in DMEM growth medium containing 4.5 g/L
Stable Transfection of HEK293 Cells with KLOTHO
Full-length human KLOTHO cDNA (3,056 bp) was cloned into a pcDNA3.1/myc-His(-) B vector in frame with a myc and 6× His tag (5,520 bp; Life Technologies) by GenScript (Piscataway, NJ). HEK293 cells were stably transfected with the expression plasmid pcDNA3.1/myc-His/KLOTHO using the PolyFect transfection reagent from Qiagen (Hilden, Germany). After transfection cells were incubated for 48 h to allow gene expression. Selection of successfully transfected cells was started by the use of Geneticin (G418 sulfate) solution (Carl Roth, Karlsruhe, Germany). Stable transfectants were selected over a time period of 4 weeks for the resistance to Geneticin. Cell clones were picked and expanded. Within the next 4 weeks, the starting concentration of Geneticin (1,000 μg/mL) was slowly reduced to 100 μg/mL. HEK293 cells that stably expressed Klotho were named HEK293-KL cells.
Fibroblast Growth Factors
Recombinant human basic FGF (bFGF; AA 1-157) was purchased from R&D Systems (Minneapolis, MN). It was Escherichia coli-derived with a predicted molecular mass of 17.4 kDa and a purity of >97%. Recombinant human FGF23 (R179Q mutant) was produced by Proteros (Martinsried, Germany) with an in-house generated HEK293 cell line stably expressing FGF23R179Q. FGF23R179Q (AA 1-251) was fused at the C-terminus to a His-tag. It showed a purity of >90% with a predicted molecular mass of 27.8 kDa.
Treatment of Cells
For induction experiments nontransfected HEK293 cells, HEK293 cells transfected with the vector control and HEK293-KL cells were treated with bFGF (10 ng/mL) or FGF23R179Q (0–100 ng/mL) for 20 min. To test small molecule inhibitors, cells were incubated with SU5402 (65 nM and 40 μM) or U0126 (100 nM and 10 μM) for 1 h and were then stimulated with bFGF (10 ng/mL) or FGF23R179Q (100 ng/mL) for 20 min. Both inhibitors were purchased from Merck (Darmstadt, Germany).
Western Blotting
Adherent cells were lysed using lysis buffer (30 mM Tris-HCl, pH 7.4, 150 mM NaCl) containing protease (Complete; Roche, Mannheim, Germany) and phosphatase inhibitor cocktails (PhosStop; Roche). Ten or 20 μg of whole cell lysate was subjected to 4–20% or 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (Immobilon-P; Millipore, Billerica, MA). Roti-Block reagent (Carl Roth) was used for blocking. The membranes were incubated with the following primary antibodies (Ab): anti-Klotho polyclonal goat Ab (1:200; Santa Cruz Biotechnology, Dallas, TX), anti-p-extracellular signal-regulated kinase 1 and 2 (ERK1/2) monoclonal rabbit Ab (1:1,500; Cell Signaling Technology, Danvers, MA), anti-ERK1/2 monoclonal rabbit Ab (1:1,000; Cell Signaling Technology), or anti-His(C-term) Ab (1:5,000; Life Technologies). After incubation with the corresponding secondary antibody signals were visualized with the enhanced chemiluminescence system (Amersham ECL Plus Western blotting detection system; GE Healthcare, Chalfont St. Giles, United Kingdom).
AlphaScreen® SureFire® p-ERK1/2 Assay
The AlphaScreen SureFire p-ERK1/2 assay from PerkinElmer (Waltham, MA) was used to measure the phosphorylation of ERK1/2 in cellular lysates. HEK293 and HEK293-KL cells were washed with 1× phosphate-buffered saline, trypsinized, and resuspended to a density of 260,000 cells/mL in cell culture medium. The cell suspension (13,000 cells per well; 50 μL per well) was dispensed into white 384-well plates (CulturPlate; PerkinElmer) and incubated (37°C; 5% CO2) overnight. The next day cells were pretreated either with compound (1 mM stock solution) dissolved in 100% dimethyl sulfoxide (DMSO) or DMSO alone. For IC50 determination compounds were diluted in 100% DMSO (ten concentrations, 0.2–80 μM). Then, 0.4 μL of compounds/DMSO were transferred to 50 μL cell culture medium per well to keep the final DMSO volume concentration at 0.8%. The cells were then incubated (37°C; 5% CO2) for 1 h before adding 13 μL FGF23R179Q (485 ng/mL) or medium to each well. The plate was again incubated (37°C; 5% CO2) for 20 min. Next, medium was removed and cells were lysed with 8 μL of AlphaScreen SureFire 1× lysis buffer per well. Lysis took place at room temperature for 10 min with gentle shaking (phmp-4 thermoshaker for microplates; Grant Instruments, Shepreth, UK). Subsequently, the reaction mix (9 μL) was added to the lysed cells followed by gentle shaking for 2 min. The reaction mix consisted of reaction buffer, activation buffer, acceptor and donor beads (in a 2:1 ratio) prepared under low light conditions. The plate was incubated 2 h at room temperature in the dark. Finally, plates were read on the EnVision® Multilabel Reader (PerkinElmer) and data analysis was done with GraphPad Prism statistical analysis software using the nonlinear regression for a sigmoidal dose–response with a variable slope (Table 1). The Z′ factor was calculated as described by Zhang et al. 24
Alphascreen Surefire Protocol to Measure FGF23-Induced ERK1/2 Phosphorylation
1. Solid white tissue culture-treated 384-well plates.
3. Compound library is dissolved in DMSO (1 mM stock solution).
5. FGF23R179Q is dissolved in phosphate-buffered saline (0.8 mg/ml stock solution) and is further diluted with medium.
10. Reaction mix consists of reaction buffer, activation buffer (7-fold dilution), acceptor beads (70-fold dilution), and donor beads (140-fold dilution); beads are added under low light conditions.
13. PerkinElmer EnVision Multilabel Reader with standard AlphaScreen option (Em = 570 nm).
DMSO, dimethyl sulfoxide; ERK1/2, extracellular signal-regulated kinase 1 and 2; FGF23, fibroblast growth factor 23; FGF23R179Q, FGF23, R179Q mutant; HEK293-KL cells, human Klotho in HEK293 cells.
Homogenous Time-Resolved Fluorescence HTRF® p-ERK Cellular Assay
The phosphorylation status of ERK1/2 in cell lysates can also be determined by the homogenous time-resolved fluorescence (HTRF) p-ERK cellular assay from Cisbio (Codolet, France). As with the AlphaScreen SureFire assay procedure, 13,000 HEK293 or HEK293-KL cells per well were cultured in 50 μL growth medium in white 384-well plates (CulturPlate; PerkinElmer) for 24 h and were then treated with compounds for 1 h before adding 13 μL FGF23R179Q (485 ng/mL) or medium to each well for 20 min. Next, medium was removed and cells were lysed with 25 μL of 1× lysis buffer containing blocking reagent. Lysis took place at room temperature for 30 min with gentle shaking (phmp-4 thermoshaker for microplates). Afterward, 16 μL of the cell lysate of each well were transferred to a new 384-well plate (ProxiPlate; PerkinElmer) and 4 μL of the premixed antibody solutions (in a 1:1 ratio) were added followed by gentle shaking for 2 min. The plate was incubated 2 h at room temperature. Finally, HTRF emissions were measured at 620 and at 665 nm on the EnVision Multilabel Reader. Emissions at 620 nm were used as an internal reference, while emissions at 665 nm were used as an indicator of the biological reaction being assessed. Within ratiometric reduction of data, the so-called HTRF Ratio (HTRF emission 665nm/HTRF emission 620nm ×104) was calculated. Data analysis was done with GraphPad Prism statistical analysis software using the nonlinear regression for a sigmoidal dose–response with a variable slope.
Cell Viability Test
To investigate the toxicity of small molecule compounds on HEK293-KL cells, the CellTiter-Blue™ (CTB) cell viability assay (Promega, Madison, WI, USA) was utilized. HEK293-KL cells were cultured in 384-well plates (13,000 cells per well; 50 μL per well) for 24 h. The next day cells were treated with 0.4 μL of small molecules (1 mM stock solution) dissolved in DMSO or with 0.4 μL DMSO alone per well and incubated (37°C; 5% CO2) for 1 h. Subsequently, 10 μL of CTB reagent was directly added to each well. The plates were incubated (37°C; 5% CO2) for 1 h before reading on the EnVision Multilabel Reader (PerkinElmer). Viable cells are able to reduce the indicator dye used in the assay, which results in a fluorescent signal. Nonviable cells rapidly lose metabolic capacity and thus do not or less generate any fluorescence.
Automation
Plate and liquid handling was performed using a HTS platform system composed of a Sciclone G3 Liquid Handler with a Twister II Robotic Arm and Flexdrop Dispenser from PerkinElmer, a MultiFlo™ Dispenser (Biotek Instruments, Bad Friedrichshall, Germany) and a Cytomat™ Incubator (Thermo Fisher Scientific, Waltham, MA, USA). AlphaScreen SureFire and CTB measurements were performed using an EnVision Multilabel Reader (PerkinElmer). Assays were carried out in white CulturPlates-384 (PerkinElmer). The plates were coated with poly-D-lysine (Sigma-Aldrich, St. Louis, MA, USA) to facilitate a better cell adherence. The diverse small molecule library used in HTS was acquired from Prestwick Chemical (Illkirch, France). The Prestwick Chemical library contains 1,280 small molecule compounds, which are 100% approved drugs (Food and Drug Administration [FDA], European Medicines Agency [EMA] and other agencies). The purity of the compounds was >90% as reported by the provider of the compounds.
Other Reagents
All other reagents not listed above (e.g., buffers) were purchased from Sigma-Aldrich (Taufkirchen, Germany) or Roth (Karlsruhe, Germany) and were of highest quality.
Results
FGF23/FGFR1c/Klotho Cell Line Development
Urakawa et al. have shown that FGF23 binds to the receptor complex FGF receptor 1c (FGFR1c)/Klotho and activates the mitogen-activated protein kinase (MAPK) signaling cascade resulting in the phosphorylation of mitogen-activated protein kinase kinase 1 and 2 (MEK1/2) and ERK1/2 in addition to an increased expression of downstream targets, for example, EGR1 (Fig. 1). They could demonstrate that the co-receptor Klotho is essential for endogenous FGF23 function by converting the canonical FGF receptor into a specific receptor for FGF23 ligand. 25 While others published cell lines overexpressing murine proteins like mouse Klotho and diverse mouse FGFR fragments, 26 we established a cell-based system with human Klotho and FGFR for the identification of small molecules interfering with the FGF23-dependent MAPK signaling pathway. Although the similarity between human and mouse proteins is usually very high, we decided to focus on a human cell-based system to closely reflect the situation in patients. With HEK293 human embryonic kidney cells, a cell line was chosen that endogenously expresses FGFR1c, while no Klotho expression was detectable (Fig. 1 and Fig. 2A). Thus, HEK293 cells were stably transfected with an expression plasmid encoding the full-length human KLOTHO. Cell clones were selected, expanded, and the expression of Klotho was confirmed by western blot analysis using antibodies against human Klotho (Fig. 2A). HEK293 cells that stably expressed Klotho were designated as HEK293-KL cells.
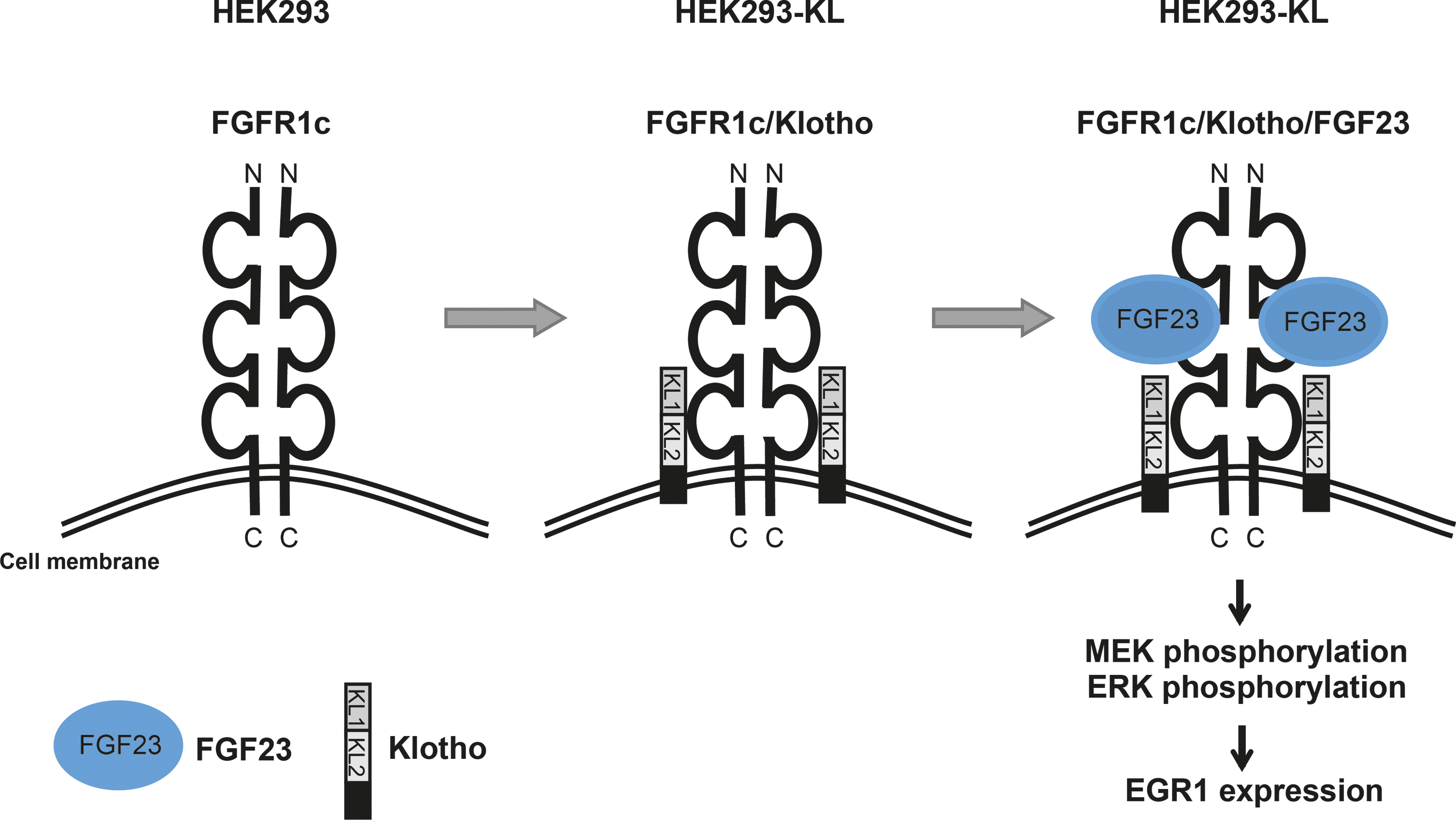
Schematic model of fibroblast growth factor 23 (FGF23)-inducible HEK293 cells stably expressing Klotho (HEK293-KL cells).
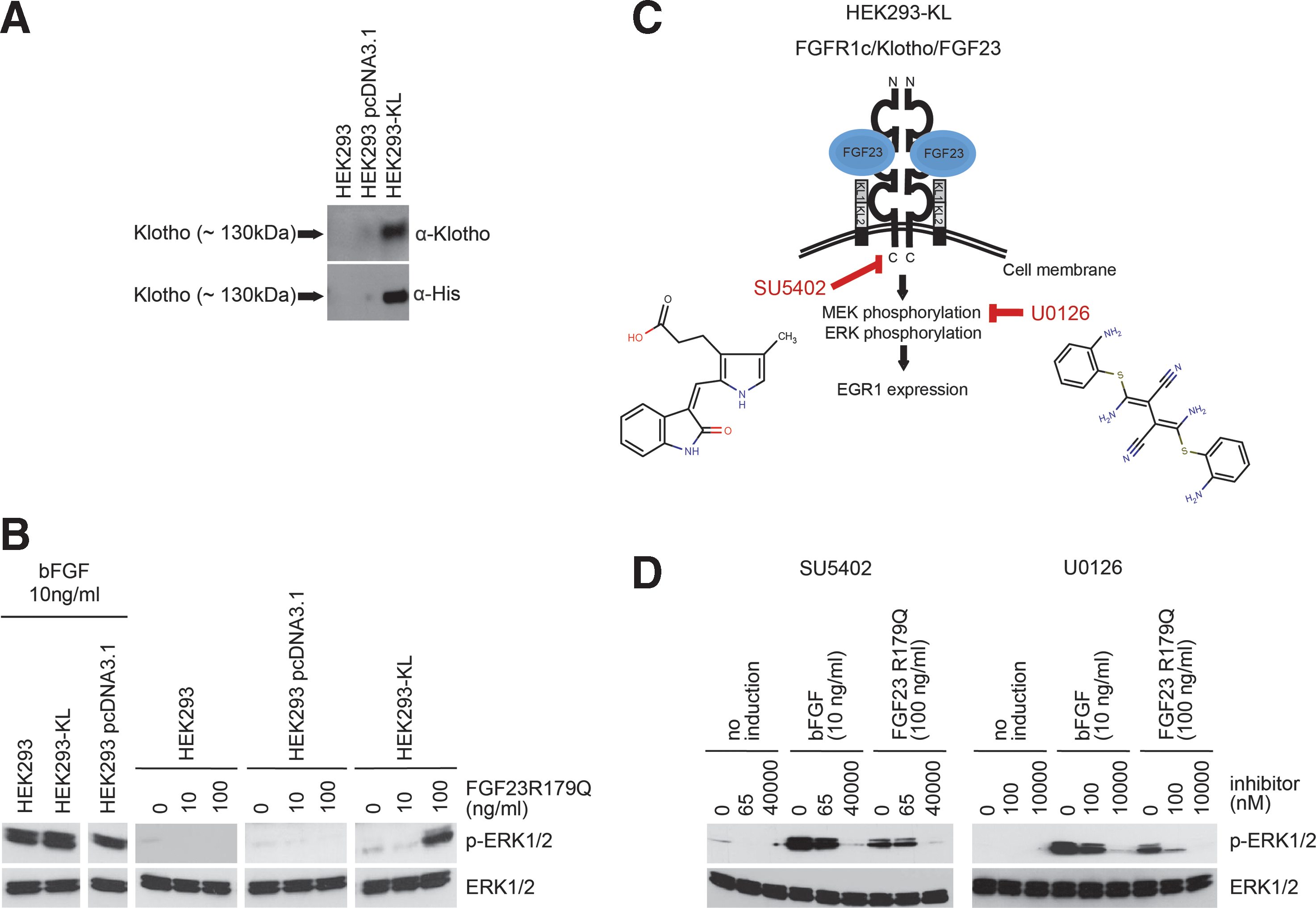
Construction and characterization of the HEK293-KL cell line.
To test whether our obtained HEK293-KL cell clones are FGF23-inducible, induction experiments were performed using a mutant form of human recombinant FGF23 (FGF23R179Q), which is known to be resistant to proteolytic cleavage, however, is fully active in inducing FGFR1c/Klotho signaling and therefore is broadly used as a tool to study FGF23 signaling. 27 As a positive control, human recombinant bFGF was used, which binds with high affinity to all four FGFRs without the need of the co-receptor Klotho. 28,29 Nontransfected HEK293 cells, HEK293 cells transfected with the vector control and HEK293-KL cells were treated with bFGF (10 ng/mL) or FGF23R179Q (0–100 ng/mL) for 20 min. All three cell lines could be activated by bFGF as evidenced by the formation of phosphorylated ERK1/2 (p-ERK1/2) (Fig. 2B). In contrast, only HEK293-KL cells expressing KLOTHO were inducible by FGF23R179Q and led to p-ERK1/2 formation (Fig. 2B). The titration of FGF23R179Q using HEK293-KL cells indicated a final FGF23R179Q concentration of 100 ng/mL being necessary for sufficient activation of the signaling pathway and thus this concentration was chosen for subsequent experiments, especially as this concentration reflects the pathological situation in patients. 30 –32
To study whether the induction of HEK293-KL cells with FGF23R179Q can be reduced by small molecule compounds, the effect of two known inhibitors of MAPK signaling pathway was investigated: SU5402 and U0126 (Fig. 2C). 33 SU5402 is a cell-permeable, reversible, and ATP-competitive inhibitor of the FGFR1 tyrosine kinase activity by interacting with its catalytic domain. 34 U0126 is a potent and specific inhibitor of MEK1/2. 35 HEK293-KL cells were incubated with SU5402 or U0126 in serum-free medium for 1 h and were then stimulated with bFGF (10 ng/mL) or FGF23R179Q (100 ng/mL) for 20 min. Concentrations of SU5402 were 65 nM and 40 μM, concentrations of U0126 were 100 nM and 10 μM. Both inhibitors reduced the activation of MAPK signaling induced by bFGF or FGF23R179Q in a concentration-dependent manner (Fig. 2D). Taken together, these results clearly demonstrate that the FGF23-inducible HEK293-KL cell line has been successfully established and that this cell model is a suitable tool to investigate the effect of small molecule compounds on FGF23-specific signaling.
Establishment of an HTS Compatible Cell-Based Assay
Next, our cell-based assay was adapted to the AlphaScreen SureFire p-ERK1/2 technology from PerkinElmer. It allows the detection of p-ERK1/2 in cellular lysates by the formation of in-solution sandwich antibody complexes, which are captured by AlphaScreen donor and acceptor beads. This triggers a cascade of energy transfer from donor to acceptor beads via singlet oxygen resulting in light emission. The intensity of light emission, referred to as Alpha signal, is proportional to ERK1/2 phosphorylation. We used this assay format to monitor the activation of MAPK pathway in FGF23-stimulated HEK293-KL cells. To make the primary assay suitable for HTS approaches, a number of important configurations had to be optimized. First, cell titration experiments were performed to identify the optimal cell numbers for the 384-well format. Adherent HEK293-KL cells were seeded at cell densities between 5,000 cells/well and 20,000 cells/well and were cultivated at 37°C in a cell culture incubator for 24 h. At a cell density of 13,000 cells/well, cells grew in a confluent monolayer, which was optimal for the assay (data not shown). When confluent, many signaling pathways like MAPK signaling pathway can become quiescent and synchronized. 36 Thus, after stimulation with FGF23 the cells can respond uniformly. Once the cell number had been defined, optimal agonist concentration (FGF23) and stimulation time were determined. Therefore, HEK293-KL cells were stimulated with increasing concentrations of FGF23R179Q for 20 min (Fig. 3A). We obtained an EC50 value of ∼110 ng/mL. According to our previous results from induction experiments, a concentration of 100 ng/mL appeared to be appropriate to create a big signal window (SW). Again, we kept the concentration of 100 ng/mL as this reflects the pathological situation. 30 –32 In the next step, the stimulation time was investigated. HEK293-KL cells were treated with 100 ng/mL FGF23R179Q for 20, 40, and 60 min. Significant changes in the phosphorylation status of ERK1/2 were detectable in cell lysates after stimulation at all time points in a comparable range. Already after 20 min the cells were strongly stimulated and the signal remained stable over time (Fig. 3B). To identify small molecules, which act in the very early phase of the FGF23-dependent MAPK signaling and to avoid effects that were produced due to feedback loops in the signaling cascade, we chose a stimulation time of 20 min for further experiments. In addition, we could confirm that our assay detected an increase of the Alpha signal after FGF23R179Q induction only in HEK293-KL cells and not in the parental HEK293 cell line. Without FGF23R179Q, just a background signal (basal activation of MAPK pathway) was detectable in both HEK293 and HEK293-KL cells (Fig. 3C).
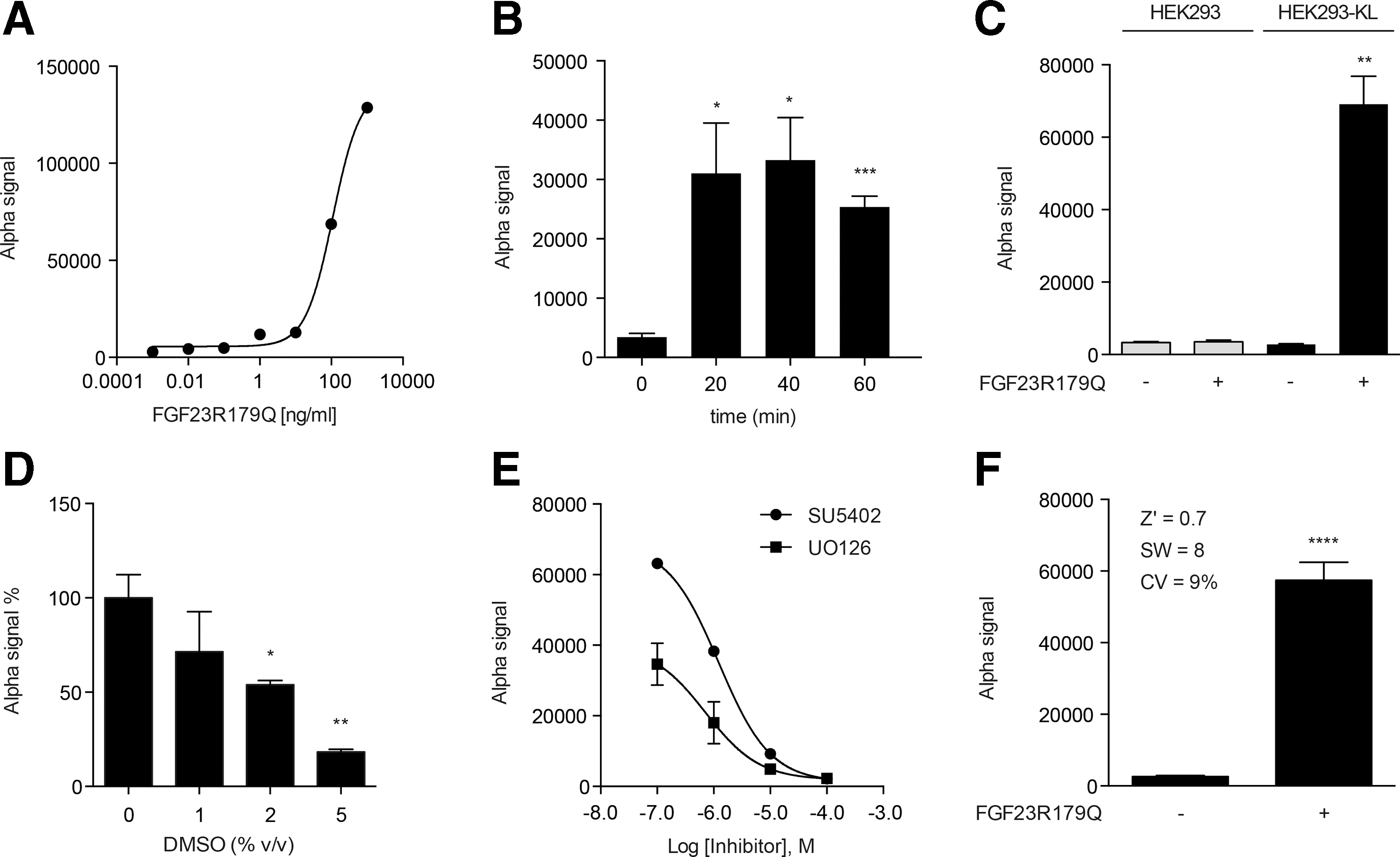
Establishment of the primary p-ERK1/2 AlphaScreen SureFire assay.
DMSO is routinely used as a solvent for compound libraries including ours. To check whether DMSO had an effect on the assay performance by reducing signal maximum, HEK293-KL cells were preincubated with a final volume concentration of 0%, 1%, 2%, and 5% DMSO for 1 h and subsequently stimulated by FGF23R179Q (100 ng/mL) for 20 min. The assay demonstrated acceptable DMSO tolerance up to 1% DMSO (Fig. 3D). In this study, screenings were carried out with a final DMSO volume concentration of 0.8% ensuring little DMSO influence on assay performance.
Inhibitor IC50 Determination and Statistical Validation for HTS Application
To validate the applicability of the developed assay for identifying small molecule compounds that modulate the effect of FGF23 signaling, the performance of the assay was tested with the known small molecule inhibitors SU5402 and U0126. HEK293-KL cells were plated in 384-well plates, treated with increasing inhibitor concentrations (0.1–100 μM) for 1 h, and then stimulated with FGF23R179Q (100 ng/mL) for 20 min (Fig. 3E). We obtained a concentration-dependent reduction of the Alpha signal with IC50 values of 1.23 μM (SU5402) and 0.72 μM (U0126), respectively, suggesting that our assay is perfectly well suited for compound screening. Finally, we tested the assay protocol under automation to determine the robustness and the throughput of the assay. We successfully adapted all dispensing and incubation steps using a liquid handling workstation and robotics. Additionally, test experiments were conducted to determine inter-plate and inter-day variations. We assessed the suitability of our assay for HTS by calculating performance measures: Z′ factor, SW and coefficient of variation (CV). All criteria exceeded the minimum pass criteria for HTS: Z′ >0.5, SW >2 and CV <20% 24,37,38 indicating an excellent assay performance and optimal robustness (Fig. 3F).
Screening of 1,280 FDA-Approved Drugs
In a first screening we intended to yield important information about the anticipated number of hits and robustness of the established assay. Moreover, since serum levels of the phosphaturic factor FGF23 are elevated in most hereditary hypophosphatemic disorders, 39,40 we were highly interested in identifying small molecule compounds with an inhibitory action on FGF23 signaling. The Prestwick Chemical library was employed as a small set of compounds for first screening. This library contains 1,280 small molecules, which are 100% approved drugs (FDA, EMA, and other agencies). Consequently, it presents a high degree of drug-likeliness.
We conducted the screening using our developed primary AlphaScreen SureFire cell-based assay. HEK293-KL cells were seeded in 384-well plates and incubated with the small molecule compounds at a final concentration of 8 μM (n = 1) for 1 h and then induced with FGF23R179Q (100 ng/mL) for 20 min. In parallel, toxicity of all 1,280 compounds on HEK293-KL cells was examined with the CTB cell viability assay from Promega. During the automated screening process the assay fulfilled all appropriate quality criteria. The p-ERK1/2 Alpha signal of the negative and positive control remained very stable over all four screening plates (Fig. 4A). An overall Z′ factor value of 0.62 and an overall SW value of 5.6 indicated an excellent assay performance and robustness (Fig. 4B). The classification of actives was based on compounds that yielded a decrease in the Alpha signal >40% (Fig. 4C, left). However, this population would include false positives that arise by affecting the AlphaScreen technology. Therefore, data obtained from the screening were compared to the results of three other unrelated AlphaScreen campaigns to identify potential frequent hitters (FHs). FHs are false positive hits, which are produced for example, by optical interference or inhibition of the detection system. 38,41 The three AlphaScreen HTS assays selected for FH identification made use of protein–protein interaction (PPI) assays from unrelated signaling pathway targets and presented excellent and robust signals (Z′ >0.5) (data not shown). The specific PPI protein pairs were screened against the same FDA-approved drugs. Therefore, combinatorial analysis of all four screening campaigns allowed the identification of FHs reducing Alpha signals independent of target selectivity. Actives were classified as FHs when they yielded a mean decrease in normalized signal >15% in all three unrelated AlphaScreen campaigns (Fig. 4C, left). Using this evaluation method, nine primary hits were classified as FHs and 41 compounds as actives (green population in Fig. 4C, left). To discriminate between nontoxic and toxic actives the results of all actives were aligned with the data of the CTB assay (Fig. 4C, right). After data analysis, 16 molecules were defined as toxic actives by reducing CTB signal by >20% (in red).

Summary of the first high-throughput screening (HTS) screen (4 × 384-well plates).
In summary, 25 remaining hits reduced the p-ERK1/2 Alpha signal by more than 40%, did not affect signals in other AlphaScreen campaigns, and showed low toxicity in the CTB assay (CTB signal >80%). Therefore, these hits were nominated as nontoxic actives representing a final hit rate of ∼2%.
Small Molecule Validation
The 25 actives were reordered as powder stocks and prepared for serial dilutions. The efficacy of all compounds was determined on the primary AlphaScreen SureFire assay in triplicates using 10-point titrations (0.2–80 μM). These experiments should confirm the initial screening results and deliver IC50 values for the individual hit compounds. Out of 25 compounds 8 showed no activity after retesting (data not shown) and thus were immediately discarded. The remaining 17 compounds showed dose-dependent inhibition of FGF23 signaling to various degrees (Fig. 5A and Supplementary Fig. S1). Subsequently, we selected the most potent inhibitors within this primary panel of compounds for retesting in a secondary assay. Out of 17 compounds 8 reduced the Alpha signal by >80% while having IC50 values <25 μM. To confirm their efficacy, an independent secondary assay was established (data not shown). This secondary assay was based on the HTRF p-ERK cellular assay format from Cisbio, which also measures the phosphorylation of ERK1/2 in cellular lysates, but makes use of a different technology for detection. In brief, it involves two diverse specific antibodies, one labeled with Eu3+-cryptate (donor) and the second with d2 (acceptor). When the dyes are in close proximity, the excitation of the donor with a laser triggers a time-resolved fluorescence resonance energy transfer (TR-FRET) toward the acceptor. The signal intensity is proportional to ERK1/2 phosphorylation. Within ratiometric reduction of data, the so-called HTRF Ratio was calculated. For the secondary assay no automation step was required, since the number for secondary retesting was limited. By the use of this independent secondary readout format we again profiled the identified 8 compounds in 10-point titrations (Supplementary Fig. S2). For seven out of eight hits we obtained inhibition of ERK1/2 phosphorylation with an IC50 < 25 μM. We ultimately retested the seven hits (active in Alpha Surefire and HTRF assays) for their inhibitory action on FGF23/FGFR1c/Klotho signaling using our p-ERK1/2 western blot assay as a tertiary readout (Fig. 4D). Importantly, the western blot analysis provides a completely different readout and allows a more direct examination of the p-ERK1/2 signal. Therefore, HEK293-KL cells were treated with increasing compound concentrations (0–30 μM) of the seven remaining compounds and additionally stimulated with FGF23R179Q (100 ng/mL). To estimate the impact on the signaling, total levels of ERK1/2 and p-ERK1/2 signal were detected with highly specific antibodies in western blot. In total, three out of seven hits showed no inhibitory effect on p-ERK1/2 (Supplementary Fig. S3). The other four hits (Pimozide, Lomerizine hydrochloride, Flunarizine dihydrochloride, and Tioconazole) led to a dose-dependent reduction in the p-ERK1/2 signal (Fig. 5B) and their structures are illustrated in Figure 5C. The total ERK1/2 signal remained unchanged, indicating that these compounds do not generally reduce ERK1/2 protein levels. Importantly, none of the four molecules showed toxic effects at the tested concentrations (data not shown) as expected, because only nontoxic compounds have been selected initially.
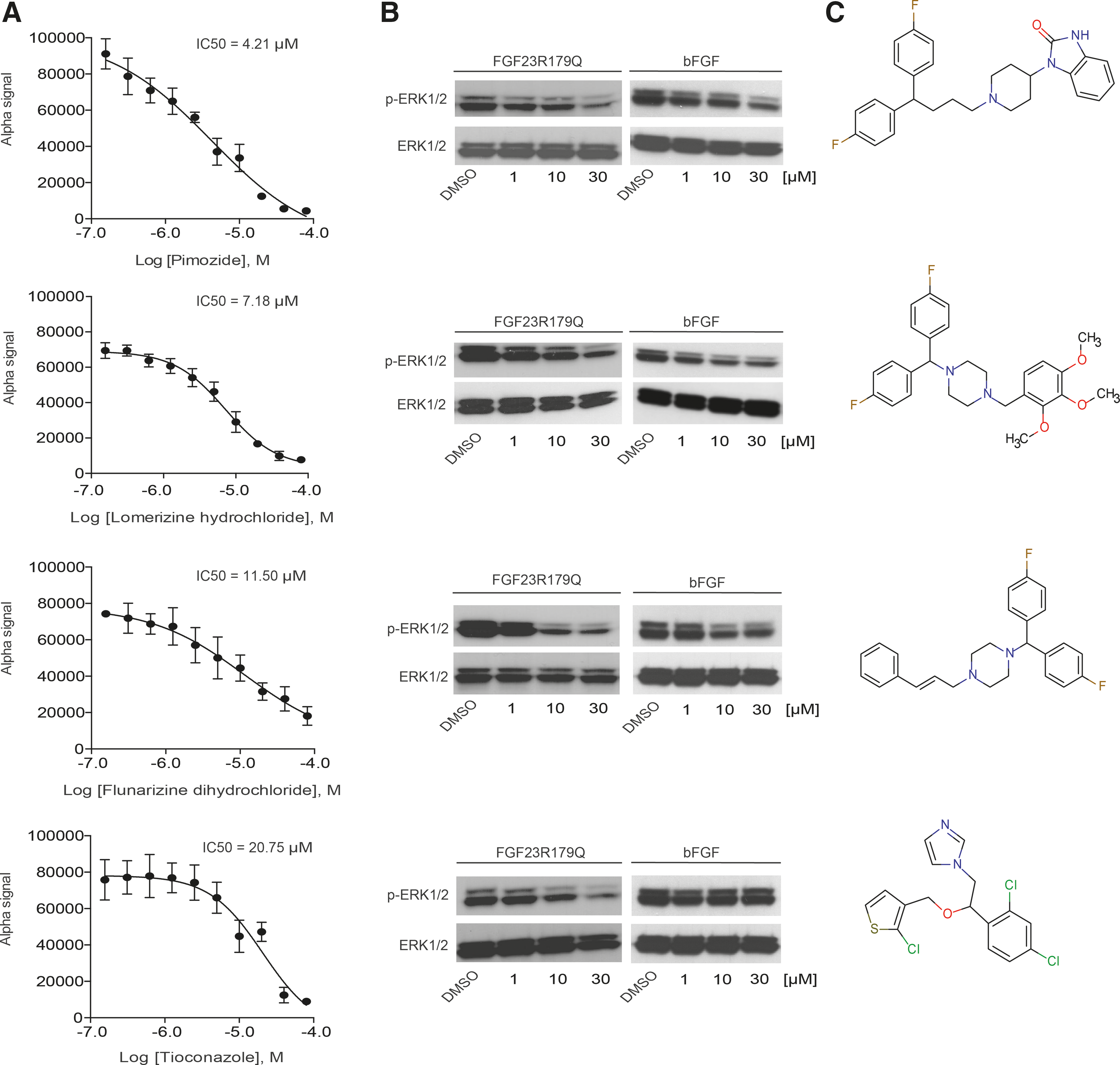
Hit validation results of four selected hits.
Taking into consideration that 1,280 small molecules were investigated in the screening, the validated hit number of four compounds after dose–response validations in Alpha SureFire and HTRF assays together with western blot analysis denotes a final validated hit rate of 0.3%. All four remaining hits showed significant reduction of p-ERK1/2 signal in three different assay formats. Of important note, the reduction of the signal was not due to general toxic effects of the molecules on HEK293-KL cells, but rather by affecting MAPK signaling.
Finally, we investigated whether the identified compounds are specific FGF23 modulators without affecting other FGFs. In a counterscreening approach, HEK293-KL cells were treated with Pimozide, Lomerizine hydrochloride, Flunarizine dihydrochloride, or Tioconazole (0–30 μM) for 1 h and were then induced with bFGF (10 ng/mL) for 20 min followed by p-ERK1/2 western blot analysis (Fig. 5B). Three compounds (Pimozide, Lomerizine hydrochloride, and Flunarizine dihydrochloride) reduced ERK1/2 phosphorylation levels after bFGF stimulation in a comparable manner like after FGF23 stimulation. Interestingly, the small molecule Tioconazole did not inhibit the activation of MAPK signaling after bFGF induction, while FGF23-mediated phosphorylation of ERK1/2 was clearly reduced.
Discussion
This report describes the development of a highly specific cell line (HEK293-KL cells) and the establishment of an HTS assay format to screen for small molecule inhibitors of FGF23/FGFR1c/Klotho receptor signaling. The HEK293-KL cell line presented in this study was carefully characterized for Klotho expression and stimulation-dependent downstream signaling. As a consequence of Klotho expression only the modified HEK293-KL cell line reacted to treatment with FGF23. The original HEK293 cell line without any detectable Klotho expression was not inducible by FGF23 treatment. In contrary, bFGF stimulated HEK293 and HEK293-KL cells to the same extend, which demonstrates that we created a cell line that is highly specific and sensitive to FGF23 stimulation. Importantly, the FGF23 used for stimulation was highly pure and purified from human cell culture expression systems. The HEK293-KL cell line was subsequently used to adopt it to the HTS p-ERK1/2 AlphaScreen SureFire assay format from PerkinElmer. The assay exceeded the minimum criteria (i.e., Z′ >0.5 and SW >2) and met the general requirements for HTS compatibility such as acceptable DMSO tolerance up to 1%. Moreover, the assay shows similar sensitivity to known inhibitors when compared with the results of western blot analysis. The results reproducibly demonstrate the more potent inhibition by U0126 compared to SU5402 in both assay types. Beside the establishment of the HEK293-KL cell line and the HT screening assay this study provides a straightforward strategy to identify FHs created by interference with the assay technology and to exclude toxic events. Compounds can generate nonspecific effects on AlphaScreen technology by singlet oxygen quenching, color quenching, and auto-fluorescence and thus reduce assay signals. Furthermore, toxic molecules can reduce the number of living cells in the well and thereby decrease the p-ERK1/2 assay signal independent of direct effects on MAPK signaling. With this strategy we scaled the number of 50 primary hits down to 25 nontoxic and specific actives. This outcome highlights the importance to identify FHs and toxic compounds for any assay readout to avoid wrong interpretation of HTS primary data sets.
In the following, we reanalyzed the efficacy of 25 actives, while only 17 compounds demonstrated dose-dependent activity. Here, 8 out of 17 molecules gave rise to sufficient inhibitory effects (>80% reduction of the Alpha signal) and IC50 values <25 μM. These 8 compounds were selected for retesting on FGF23/FGFR1c/Klotho signaling in an independent secondary assay based on a different detection technology. Seven out of eight compounds showed IC50 values <25 μM (Fig. 4D). Eventually, a dose-dependent reduction of ERK1/2 phosphorylation was revealed for four out of these seven compounds using our tertiary western blot test system. Importantly, we used strict criteria for our hit selection as our validated hits should demonstrate sufficient inhibitory potential in all three assay formats. Pimozide, Lomerizine hydrochloride, and Flunarizine dihydrochloride are members of a class of diphenyl-methyl or -butyl amins that are known to have functions in many different indications ranging from schizophrenia to migrainous vertigo. 42 –44 Tioconazole is an antifungal drug of the imidazol antimycotics class. 45 Up till now, no interaction of the identified hit compounds with FGF23/FGFR1c/Klotho signaling or MAPK signaling in general has been described. Interestingly, the three diphenyl-methyl or -butyl amins (Pimozide, Lomerizine hydrochloride, and Flunarizine dihydrochloride) target receptors and channels, respectively, thereby assuming a preference for membrane associated actions. In a first functional study we aimed to determine whether these compounds are FGF23-specific without affecting another FGF (bFGF) and whether they directly interact with the FGFR1c/Klotho receptor complex or influence the MAPK signaling pathway downstream of the receptor. Specific actions on the FGFR1c/Klotho receptor would proof the feasibility of directly targeting this receptor for generation of highly specific FGF23 antagonists. Previous studies have revealed that small molecules that target either FGFR tyrosine kinase activity (PD173074 and NVP-BGJ398) 19,20 or inhibit downstream FGF23 signaling (PD0325901) 46 can ameliorate hypophosphatemia in animal models. Both types of interference are interesting, but interaction with the unique FGF23 receptor complex is expected to be more specific than inhibition of MAPK pathway as this is a major signal cascade from the cell surface to the nucleus involved in miscellaneous biological functions. 47 Thus, early counterscreening of validated hits against bFGF is most important to extract FGF23-specific inhibitors and discard general MAPK/ERK modulators that may harbor adverse effects. In addition to their detected inhibitory effect on FGF23 signaling, the three structurally related compounds Pimozide, Lomerizine hydrochloride, and Flunarizine dihydrochloride reduced the phosphorylation status of ERK1/2 also after the induction with bFGF, suggesting that they are rather general modulators of the downstream MAPK/ERK cascade. Tioconazole, however, only reduced the level of p-ERK1/2 in HEK293-KL cells after the stimulation with FGF23 but not after the stimulation with bFGF. This fact is intriguing with regards to selective interference of Tioconazole with the FGF23/FGFR1c/Klotho signaling pathway and further functional studies are now warranted to characterize the specific inhibitory nature of this validated hit compound.
In summary, we successfully established a cell line that we could adopt for HTS campaigns to screen for small molecules interfering with the FGF23/FGFR1c/Klotho-dependent MAPK signaling. We were able to screen a collection of 1,280 compounds and provide a strategy for identifying nontoxic and specific actives. Future efforts will focus on (i) elucidating the mode of action of the current validated hit set for better profiling toward proof of concept and (ii) on screening larger collections of drug-like compounds to expand the pool of active small molecules for developing FGF23-specific antagonists.
Footnotes
Acknowledgments
This work was supported by the German Ministry of Education and Research BMBF OSTEOPATH grant (01EC1006B). We thank Scarlett Dornauer for excellent technical assistance.
Disclosure Statement
No competing financial interests exist.